Journal of the Mexican Chemical Society, vol. 61, no. 2, 2017
Sociedad Química de México A.C.
Jesús Campos
Universidad de Sevilla, Spain
Ernesto Carmona *
Universidad de Sevilla, Spain
Date received: 18 October 2016
Date accepted: 10 January 2017
Funding
Funding source: Spanish Ministry of Science
Contract number: CTQ2013-42501-P
Contract number: CTQ2014-51912-REDC
Contract number: CTQ-2014-52769-C3-3-R
Funding
Funding source: Junta de Andalucía
Contract number: FQM-119
Contract number: P09-FQM-4832
Contract number: FQM-2126
Funding
Funding source: EU 7th Framework Program, Marie Skłodowska-Curie actions
Contract number: 267226
Abstract.: The chemistry of late transition metal alkylidenes [M=CR2], where R is H a hydrocarbyl group, have attracted widespread attention although mainly with reference to complexes of metals in low oxidation state. We focus in this paper on reactions based on electrophilic attacks by Ph3C+ that allow either isolation of stable cationic Ir(III) alkylidenes, considerably more attractive than well-known Ir(I) counterparts, or the generation of very reactive variants that experience fast migratory insertion into existing Ir-C and Ir-H sigma bonds. The present studies are based on (η5-C5Me5)Ir(III) complexes that bear a cyclometalated PMeXyl2 ligand (Xyl = 2,6-Me2C6H3). The contribution of different monoanionic ligands (chloride, alkyl or hydride) to either stabilize the Ir=CR2 linkage or provide facile reactivity routes has been investigated, including the use of various deuterium isotopologues of the iridium complex precursors.
Keywords.: Alkylidene, Hydride abstraction, Iridium, C-C coupling, Metalacycle.
Resumen.: La química de los complejos de los metales de transición de los últimos grupos con ligantes alquilideno, [M=CR2], donde R es H o un grupo hidrocarbilo, es un área de gran interés, aunque con referencia principalmente a estados de oxidación bajos. En este trabajo se describen reacciones de ataque electrofílico mediante Ph3C+ que permiten el aislamiento de alquilidenos catiónicos de Ir(III), de mayor interés que los bien conocidos análogos de Ir(I), o alternativamente, la generación de especies muy reactivas que experimentan con rapidez reacciones de inserción migratoria en enlaces sigma Ir-C o Ir-H preexistentes. Los estudios que se describen se basan en complejos de la agrupación (η5-C5Me5)Ir(III) con el ligante PMeXyl2 ciclometalado (Xil = 2,6-Me2C6H3). Se ha investigado la capacidad de diferentes ligantes monoaniónicos (cloruro, alquilo o hidruro) para estabilizar el enlace Ir=CR2, o para proporcionar otras rutas de reacción fáciles para lo cual se han utilizado diversos isotópologos deuterados de los complejos precursores de iridio.
Palabras clave: Alquilideno, abstracción de hidruro, iridio, acoplamiento C-C, metalaciclo.
Introduction
Carbenes have been widely utilized as spectator ligands since the development of N-heterocylicarbenes (NHCs) [1] and more recently of the related cyclic (alkyl)(amino)carbenes (CAACs),[2] owing to their synthetic availability, the facility of modulating their steric and electronic properties and not least the kinetic and thermodynamic stability they confer to transition metal complexes. Together with cyclopentadienyls, tertiary phosphines and related P-donors, carbenes have become ubiquitous in transition metal chemistry including applications in homogeneous catalysis.[1] Besides, the rich reactivity of the M=C bond places metal carbenes in a prominent position in organometallic chemistry.
Carbocyclic[3] and heteroatom-stabilized carbenes (CRX; X = OR’, NR’2 and other groups)[4],[5] have been amply investigated and their reactivity comprehensively analyzed.[6],[7] A highly attractive and important group of carbenes are the so-called alkylidenes, examples of which are discussed in this article. With an ever growing number of carbene complexes of diverse reactivity, some confusion about their nomenclature has arisen. It is, therefore, pertinent to clarify that in accordance with IUPAC nomenclature, alkylidene complexes contain a M=CRR’ bond where R, R’ are hydrogen, alkyl, aryl and the like.[8] At variance with Fischer carbenes, that hold one or two heteroatoms bonded to the carbene atom, metal alkylidenes cannot be classified in a general manner as electrophilic or nucleophilic, for M=CRR’ termini of the two types exist depending on the metal, the carbene ligand and the other ligands of the complex. The capacity of metal alkylidenes to exhibit Fischer- (electrophilic) or Schrock-type (nucleophilic) reactivity has been widely exploited in homogeneous catalysis, most notably in an ample variety of olefin metathesis reactions.[9]
Although metal alkylidenes of late transition elements including iridium are typically stabilized by low oxidation states,[10],[11] over the years, our group studied the chemistry of Ir(III) complexes of hydrotris(pyrazolyl)borate ligands with carbenes of both the :CRR’ and :CRX types.[4a,b] The Ir-carbene unit was in many cases generated by double C-H bond activation reactions and we further authenticated unusual alkene-to-alkylidene reversible rearrangements involving reversible α- and β-hydrogen elimination reactions.[12] Ir(III) alkylidenes, particularly cationic variants, are highly electrophilic[13] and have been proposed as reactive intermediates for several relevant transformations,[14] including the cleavage of carbon-fluorine[15] and carbon-carbon[16] bonds.
Pursuing these studies further and following the pioneer work of Bergman and coworkers in the use of [(η5-C5Me5)Ir(Me)(PMe3)(S)]+ for the selective activation of C-H bonds[17] (S = solvent), we recently showed that (η5-C5Me5)Ir(III) complexes of dialkylxylyl phosphines, PR2(Xyl) (R = i Pr, Cy; Xyl = 2,6-Me2C6H3),[18] allowed isolation of stable cationic Ir(III) alkylidenes (structure A in Fig. 1) in reactions that required the activation of two benzylic C-H bonds.[19] Under similar conditions the related PMeXyl2 ligand afforded the cyclometalated structure B (Fig. 1) with uncommon η4-P-pseudoallyl coordination of the metalated ligand.[20] Further reactivity resulted in bicylic complex C from which α-hydride abstraction by Ph3C+ led first to the alkyl-alkylidene D and ultimately to an intriguing hydride phosphepine complex (not shown in Fig. 1) by migratory insertion of the alkylidene into the iridium alkyl bond.[20] Related Ph3C+ promoted C-C bond forming reactions have been disclosed recently by our group.[21]
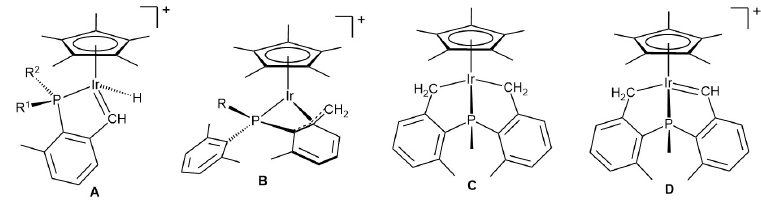
Fig. 1
Previous examples from our group of (η5-C5Me5)Ir(III) structures containing cyclometalated xylylphosphine ligands.
Such electrophilic attacks to carbon-based ligands are important reactions that occur often without cleavage of the metal-carbon bond.[22],[23] Specifically, action of Ph3C+ on a metal-alkyl bond is known to yield a metal-carbene in a process thought to occur with initial electron transfer from the metal to Ph3C+, followed by α-H atom transfer.[24] We thought of interest to broaden further recent related reactivity studies on platinum[25] and iridium 10e complexes, and in this paper we have extended these investigations, concentrating attention on the four (η5-C5Me5)Ir(III) complexes shown in Fig. 2. As discussed later, some stable cationic Ir(III) alkylidenes have been isolated, along with somewhat related hydride alkene complexes. In addition, other C-C bond coupling reactions implicating transient carbenes derived from diazo compounds[26] have been investigated.
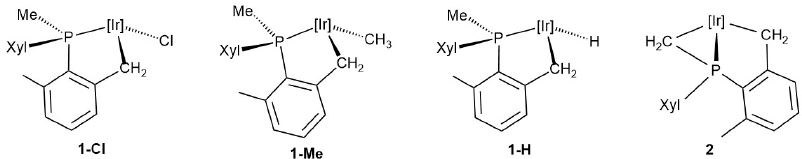
Fig. 2
Ir(III) metalacycles studied in this work [(Ir] = (η5-C5Me5)Ir)
Results and Discussion
Ph3C+promoted hydride abstraction reactions
The four Ir(III) complexes selected for this work (Fig. 2) contain a five-member phosphine-alkyl metalacycle that results from metalation of a δ benzylic bond of a PMeXyl2 ligand.[18] Chloride 1-Cl was the parent complex within this family, whereas 1-H and 1-Me are the corresponding hydride and methyl derivatives. Compound 2, that resulted from the reaction of 1-Cl with LiBu n ,[18a] is an unusual bis(iridacycle) that besides the common five member iridacycle holds an unusual three member Ir-CH2-P moiety that stems from the metalation of the P-Me bond. Clearly, as the four possess an Ir-CH2 bond within the five member ring they are prone to undergo α-H abstraction. But in addition, 1-Me and 2 bear another iridium-alkyl bond that could compete with the Ir-CH2 metalacyclic bond for electrophilic attack by Ph3C+. Moreover, complex 1-H may also experience α-hydride abstraction at the Ir-H bond, which is a common reaction of metal hydrides[22],[23] that appears to imply a single-step hydride transfer.[27]
Scheme 1 summarizes the outcome of the low-temperature reactions of complexes 1-Cl (A), 2 (B) and 1-H (C), with trityl salts, [Ph3C]+[X]- (X = PF6 or B(C6F5)4). In all cases a single reaction product was obtained though, as discussed later, formation of complex 1 + from 1-H is more complex than may be inferred from Scheme 1C. At variance with these observations, the analogous reaction of 1-Me (Scheme 2) gives a mixture of products and will be analyzed separately.
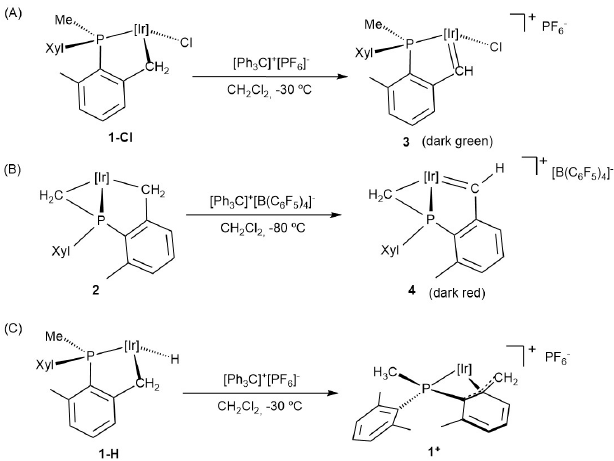
Scheme 1
α-Hydride abstraction from neutral compounds 1-Cl, 1-H and 2.
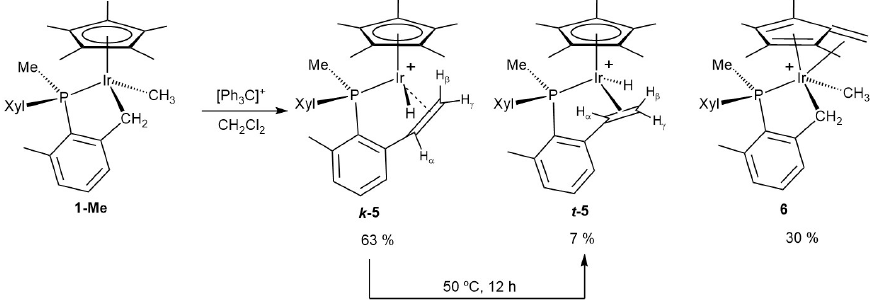
Scheme 2
Reaction of 1-Me with [Ph3C]+ and isomerization of 5.
Compounds 1-Cl and 2 underwent formal α-hydride abstraction at the Ir-CH2 bond of their five member metalacycle forming the cationic alkylidene complexes 3 and 4, respectively. No reaction of the P-bonded methylene group of 2 was detected at -80 ºC or at room temperature despite literature precedent for such reactivity.[28] Regarding the formation of the pseudo-allylic complex 1+ as in Scheme 1C, it is consistent with either direct Ir-H transfer to Ph3C+, or formation of an unobserved hydride alkylidene related to complex 3, followed by hydride attack to the highly electrophilic alkylidene carbon atom.[20b] Migratory insertion reactivity could also be expected for the cationic alkyl alkylidene complex 4. Indeed the somewhat related alkyl alkylidene D of Fig. 1 was found to undergo facile C-C bond coupling with formation of an unusual hydride phosphepine complex.[20b,c] It appears that the analogous C-C coupling in 4 is thwarted by the rigidity of its three-membered alkyl unit that would require an energetically prohibited transition state.
Solutions of compounds 3 and 4 exhibit characteristic intense green and red colors, respectively, reminiscent of our previously synthesized Ir(III) alkylidene species A and D (Scheme 1).[15,16] The thermal stability of compounds 3 and 4 is remarkable when considering prior examples of late transition metal electrophilic alkylidenes. Compounds 3 and 4 are stable under inert atmosphere in the solid state for long periods of time, and in dichloromethane solutions exhibit t1/2 of several days at 25 ºC. The two compounds are characterized (Fig. 3) by a deshielded 1H NMR alkylidene signal at 16.61 (3, d, 3JHP = 0.9 Hz) and 14.9 ppm (4, s), respectively, which are shifted by ca. 11 - 13 ppm with respect to the peaks due to the diastereotopic Ir-CH2 protons of 1-Cl and 2. The corresponding 13C{1H} signals are found at 262.4 and 243.7 (2JCP = 28 Hz) ppm, respectively, with the former featuring a one-bond C-H coupling constant of 153 Hz, in agreement with the alkylidene formulation. A marked variation in the 31P{1H} spectrum of 4 (-13.1 ppm) when compared to 2 (-41.6 ppm) is observed that is more significant than for the chloride compounds (from 11.3 in 1-Cl to 20.8 ppm in 3). Other spectroscopic data matched those of our related and previously synthesized complexes (see Experimental Section).
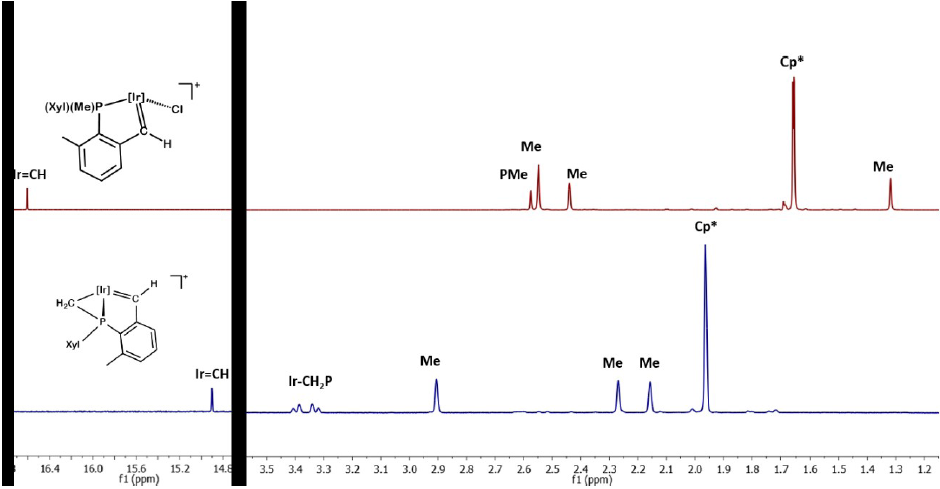
Fig. 3
1H NMR (500 MHz, CD2 Cl2 , 25 ºC) spectra of cationic alkylidenes 3 and 4.
Single crystals of 3 and 4 suitable for X-ray studies were obtained by slow diffusion of pentane into dichloromethane solutions of the complexes at -20 ºC. A characteristically short Ir1-C17 bond distance was measured for both compounds (1.899(5) Å, (3); 1.905(4) Å, (4)), that compares well to the analogous distance in compounds of type A in Fig. 1 (1.896(5) and 1.907(2) Å),[19] whereas the average Ir-C distance in our previously reported neutral and cationic alkyl complexes of similar structures is significantly longer (ca. 2.12 Å)[18b,20b,c]. As shown in Fig. 4, X-ray crystallography unequivocally confirms that hydride abstraction from 2 occurred from the Ir-CH2 methylene group within the five-membered ring, whereas the metalated P-Me group remained unaltered. The P1-C27 bond distance (1.750(4) Å) is slightly shorter than the other two P1-C bonds (ca. 1.80 Å), most likely due to the partial double-bond character of the P1-C27 unit.[29] The strain within the three-membered metalacycle is clearly reflected by the acute P1-Ir1-C27 angle of 46.84º, less than half the value that would correspond to a tetrahedral environment around the P1 atom.
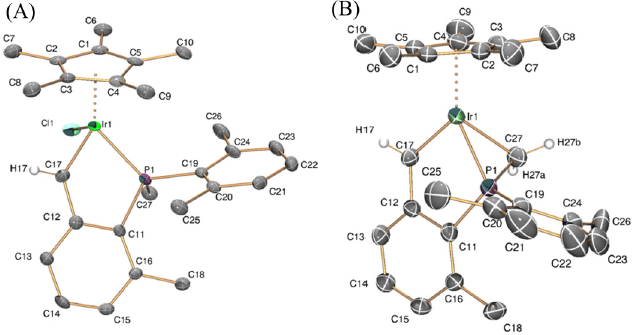
Fig. 4
ORTEP diagrams for complexes 3 (A) and 4 (B). Thermal ellipsoids are drawn at the 50 % probability. Most hydrogen atoms and counterions have been omitted for clarity.
As noted briefly, the reaction of complex 1-Me with [Ph3C][B(C6F5)4] yielded a variety of products (Scheme 2). Two of them were diastereomeric hydride alkene complexes stemming from Ph3C+-promoted C-C bond forming reactions (notice the existence of three stereogenic centers in the molecules of 5, which are chiral at Ir, P and the CH carbon of the coordinated olefin). The third product, rather oddly unobserved in the reactions of the other complexes investigated (Scheme 1), resulted from β-hydride abstraction from one of the C5Me5 methyl groups and afforded the permethylated fulvene complex 6.[30] The reaction of Scheme 2 was started at 0ºC, and after stirring at room temperature for 30 minutes afforded a kinetic mixture of isomers k-5, t-5 and 6 in a ca. 63:7:30 proportion. Since further stirring at room temperature did not change the k-5:t-5 ratio, it can be concluded that they form along independent, competitive reaction paths. As discussed later, complex k-5 derived from electrophilic attack at the Ir-CH3 bond, while t-5 resulted from an α-H abstraction reaction involving the Ir-CH2 unit. This kinetic mixture of diastereomers 5 converted into the thermodynamic isomer t-5 upon heating at 50 ºC for 12 h. 31P{1H} NMR monitoring at 40 ºC gave a first order rate constant of 1.4·10-5 s-1 (see Fig. S1), from which a ΔG≠ value of 25.3 ± 0.1 kcal·mol-1 was deduced. Related isomerization reactions of Ir(III) hydride alkene complexes have been extensively studied by our group[31] and accordingly the present system was not further investigated.
Compounds t-5 and k-5 are characterized by 1H NMR signals at -15.77 (d, 2JHP = 30.1 Hz) and -14.31 (d, 2JHP = 28.5 Hz) ppm, respectively, due to the iridium hydride group. In the 31P{1H} NMR spectra peaks at -0.1 and -9.8 ppm appear due to the thermodynamic and kinetic isomers, respectively. For the former, the olefin moiety features three 1H NMR signals at 4.27 (dd), 2.83 (dt) and 2.64 (dd) ppm, due to Hα, Hγ and Hβ, respectively. Coupling constants are in agreement with the formulation depicted in Scheme 2, with values of 10.5 and 8.6 Hz for the three-bond H-H coupling of trans (Hα-Hγ) and cis (Hα-Hβ) protons, whereas the geminal 2JHH coupling between Hβ and Hγ amounts 2.4 Hz. No coupling to the phosphorous nucleus is observed, except for Hγ, which exhibits a three-bond coupling of ca. 2.4 Hz. Corresponding 13C{1H} NMR signals appear strongly deshielded with respect to 1-Me (which exhibits resonances at 16.9 and -22.9 ppm for Ir-CH2 and Ir-CH3, respectively), with values of 62.7 (CHα) and 34.1 (CHβHγ) ppm, as expected for olefinicC13 nuclei. 1H and 13C{1H} NMR signals due to the kinetic isomer are similar to those of the thermodynamic one. The formulation depicted in Scheme 2 for the two diastereomers was tentatively inferred from 2D-NOE experiments. Compound t-5 exhibits an intense NOE cross-peak between Ir-H and P-Me, thus suggesting that both groups lie in a syn conformation. However, this cross-peak is absent in k-5, which might suggest an anti arrangement of both functionalities. As represented in Fig. 5, the structures suggested for isomers 5 were further confirmed by X-ray crystallography. These studies evidenced not only the formation of the new C-C double bond (1.470(13) and 1.473(10) Å for k-5 and t-5, respectively), but also the syn (k-5) vsanti (t-5) orientation of the Ir-H and P-Me units, without any other discernible structural differences. Other geometrical parameters are in agreement with previously discussed molecular structures of related compounds.[18-20]
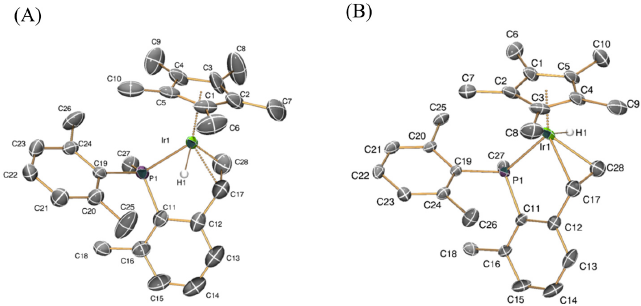
Fig. 5
ORTEP diagrams for complexes for the two diastereomers of compound 5: (A) kinetic isomer k- 5; (B) thermodynamic isomer t- 5. Thermal ellipsoids are drawn at the 50% probability. Hydrogen atoms (except Ir-H) and counterions have been omitted for clarity.
Attempts to isolate pure complex 6 were unsuccessful. The proposedformulation is therefore tentative although finds strong support on NMR data recorded for its mixtures with isomer t-5. Fig. 6 shows the 1H NMR spectrum in the region ca. 3 - 0.5 ppm for a ca. 6:4 mixture of complexes t-5 and 6 that resulted fromseveral attempts of fractional crystallization.The appearance of some distinctive signals due to the presence of a 1,2,3,4-tetramethylfulvene moiety provides strong support to the proposed formulation. In particular, olefinic resonances at 5.30 (d, 4JHP = 4.2 Hz) and 4.54 ppm attributed to the CH2 terminus, and at 1.80, 1.75, 1.57 and 0.76 ppm due to inequivalent methyl groups, each with the expected relative intensity and moreover exhibiting four-bond coupling to the 31P nucleus (1.4 - 2.8 Hz), are conclusive and comparable to literature precedents.[30]
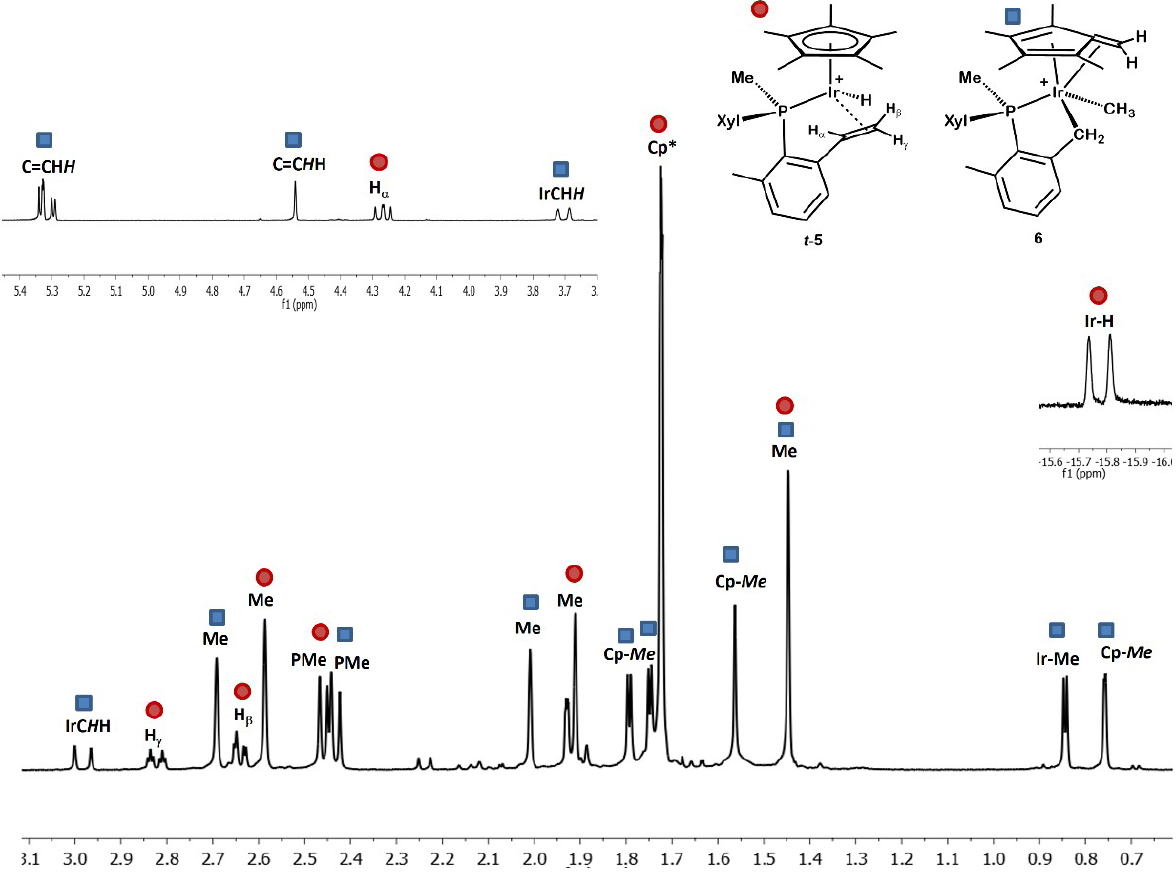
Fig. 6
1H NMR (400 MHz, CD2Cl2, 25 ºC) spectrum of a mixture of t-5 and 6 (ca. 6:4).
Mechanistic considerations. Deuterium labeling studies
As pointed out earlier, electrophilic attack by Ph3C+ to either of the neutral molecules of 1-H and 1-Me could occur at two different bonds. For hydride 1-H these are the Ir-H and one of the Ir-CH2 sites, while for the methyl analog 1-Me, α-H abstraction could occur at the Ir-CH3 and Ir-CH2 locations. To gain mechanistic insights, additional experiments were undertaken using different isotopologues of the two complexes.
Hydrogen abstraction from 1-H through either the Ir-H or the Ir-CH2 sites would clearly proceed through conspicuously different reaction routes, that would nonetheless turn into the same reaction outcome, namely complex 1+ (Scheme 1). As illustrated in Scheme 3, Ir-H abstraction would form a cationic solvento species 1S+ , known to rearrange immediately[20] to the η4-P,benzylic complex 1+ . Similarly, the Ir-CH2 path would also produce 1+ albeit with initial generation of a highly electrophilic hydridoalkylidene E, followed by a 1,2-H shift from Ir to the carbene. Isotopologues 1-D and [D11]-1-H, selectively deuterated at the hydride and all benzylic positions of the cyclometalated phosphine, respectively, were assayed (Scheme 4). The first was prepared from 1-Cl and LiAlD4 [18] whereas the second emanated from H/D exchange between 1-Cl and CD3OD [18] and subsequent reaction with LiAlH4. As delineated in Scheme 4, treatment of 1-D with 1 equiv of Ph3C+ resulted in the exclusive formation of Ph3CH, hinting faster Ir-CH2 over Ir-D reactivity. Nevertheless, the analogous experiment performed with isotopologue [D11]-1-H led to Ph3CH(D) with ca. 50% deuterium incorporation, with protio scrambling over all benzylic positions (ca. 5%). These results indicate that the two reactions have comparable rates, with H-abstraction from Ir-CH2 being somewhat faster than abstraction from the metal hydride site. Primary kinetic isotope effects mostly associated with the Ir-CH2/Ir-CD2 linkages are probably responsible for the above results.
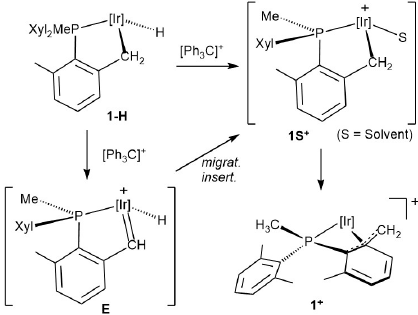
Scheme 3
Possible routes for hydride abstraction from 1-H to give 1+.
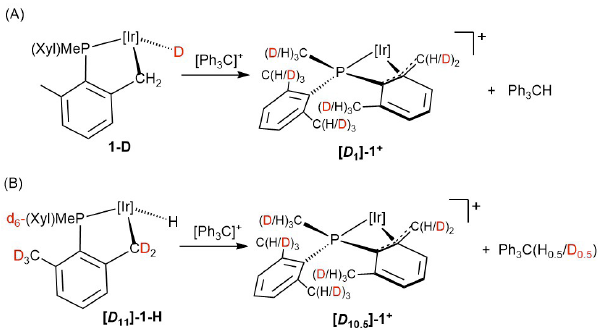
Scheme 4
Hydride abstraction by [Ph3C]+ from 1-D and [D11]-1-Hisotopologues.
Scheme 5 shows the labeled products expected from the deuterated complexes [D 11 ]-1-Me and 1-CD3 after hydride abstraction from either the Ir-CH3 (A-top and B-top) or Ir-CH2 (A-bottom and B-bottom) sites. An important aspect to keep in mind is the fact that hydride abstraction from the Ir-CH3 group would lead to a transient methylidene (Ir=CH2) that after insertion of the alkyl fragment and subsequent β-H elimination would yield the kinetic isomer of the final product k-5. In contrast, the analogous rearrangement starting with H-abstraction from the Ir-CH2 terminus would produce the diastereomer t-5.
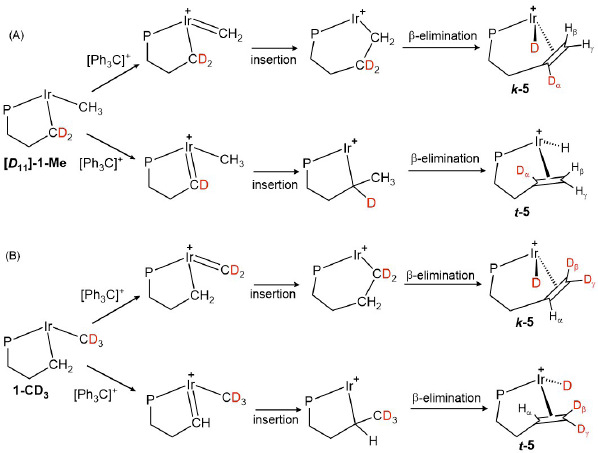
Scheme 5
Expected deuteration patterns of the products (5) resulting from Ir-CH3 or Ir-CH2 hydride abstraction of isotopologues [D 11]-1-Me (A) and 1-CD3 (B). Molecular structures have been simplified for clarty.
The distribution of the deuterium labels depicted in Scheme 6 was inferred from 1H NMR spectroscopic analysis based on the expected isotopic distribution shown in Scheme 5. These results confirmed that not unreasonably, there is a competition between abstraction of the hydride from the two alkyl positions, Ir-CH2 and Ir-CH3, with the latter, that leads to the kinetic isomer k-5, being preferred. The reactions were carried out at 25 ºC and spectroscopic analysis was acquired immediately to prevent H/D scrambling due to isomerization reactions. Primary kinetic isotopic effects were noticeable in these experiments: hydride abstraction from the protio isotopologue, 1-Me (Scheme 2) yielded a 90:10 mixture of k-5:t-5 (70% overall yield), whereas the use of [D11]-1-Me increased the ratio of the k -isomer (abstraction from Ir-CH3) at the expense of the t -isomer (abstraction from Ir-CD2), with a k:t proportion of ca. 97:3. The opposite effect was found when 1-CD3 underwent hydrogen abstraction (k:t ≈ 75:25).
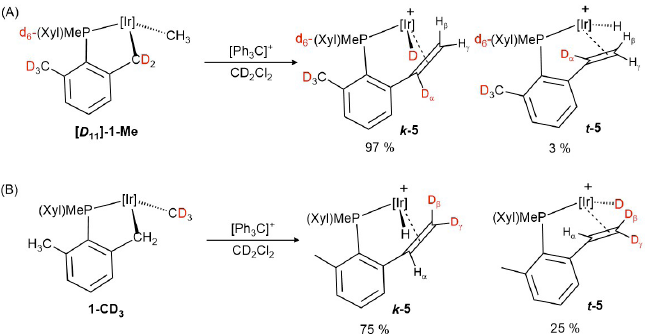
Scheme 6
Reaction of trityl cation with deuterated compounds [D11]-1-Me (A) and 1-CD3 (B).
Reaction of 1+ with Diazocompounds
As an extension of these studies we investigated the reaction of 1+ with two diazocompounds (Scheme 7) as carbene transfer reagents, with the aim of disclosing other C-C coupling reactions between alkyl and carbene fragments. Indeed, addition of either EDA (EDA = ethyldiazoacetate, N2CHCOOEt) or (trimethylsilyl)diazomethane (N2CHSiMe3) resulted in the expected C-C coupling reaction that led to complexes 7 and 8, respectively. These transformations could be regarded as models for related catalytic C-C coupling processes such as the C1 polymerization of diazocompounds[32] and other relevant transformations that involve the formation of new C-C bonds.[33] The new compounds were obtained as mixtures of cis and trans isomers of the newly formed olefinic ligand. The identification of geometric isomers was achieved with the aid of 2D-COSY and NOESY NMR experiments. A slight preference for the trans isomer was found in the formation of both 7 and 8. In fact, the proportion of trans-7 increased to ca. 75% when the reaction of 1+ with EDA was effected at -80 ºC, and then the mixture allowed to reach slowly room temperature. No intermediates were detected by low-temperature 1H NMR monitoring, but in view of the chemistry already discussed and our prior studies on related systems, the reaction involved, in all probability, highly reactive cationic alkylidene species resulting from carbene transfer from the diazocompound to the vacant site of 1+ following a change in the coordination of the benzylic terminus[20b] from η3 to η1. The resulting metal carbene subsequently rearranged by migratory insertion of the Ir-CH2 fragment and β-H elimination elementary steps.[34]
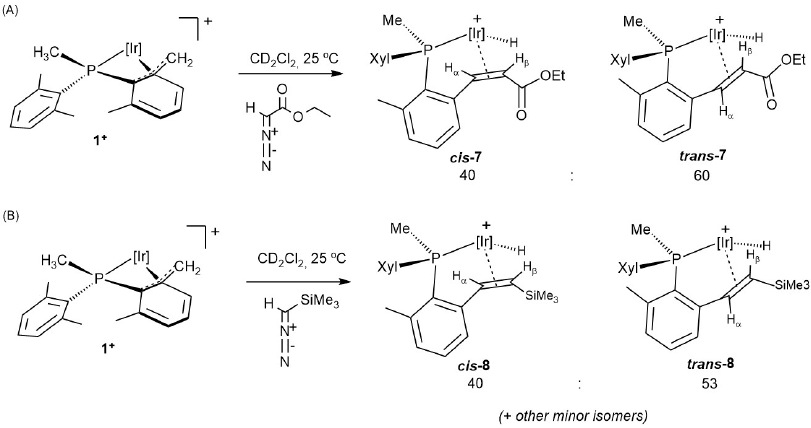
Scheme 7
Reaction of 1+ with diazocompounds EDA (A) and N2CHSiMe3 (B).
Compounds 7 and 8 exhibit strongly deshielded, highly characteristic 1H NMR doublets between -15 and -17 ppm due to the iridium hydride ligands, with a two-bond coupling to the phosphorous nucleus of around 29 Hz. Their corresponding infrared bands appear between 2150 and 2175 cm-1. Two doublets at 4.73 and 3.72 ppm in the 1H NMR spectrum of trans-7 are due to olefinic protons Hα and Hβ, respectively, and exhibit a three-bond coupling constant of 9.3 Hz. The corresponding 13C{1H} NMR signals for CHα and CHβ appear at 59.5 and 40.9 ppm, respectively, the former presenting a small coupling to phosphorous of 4 Hz. The methylene protons of the ethoxy group (OCH2CH3) are diastereotopic and give two multiplets at 4.29 and 4.14 ppm, whereas the methyl group appears as a triplet (3JHH = 7.1 Hz) at 1.32 ppm. 1H and 13C{1H} NMR data due to cis-7 are not dissimilar to those of the trans isomer (see Experimental Section for details). Similar resonances are measured for compounds 8. The olefinic protons of cis-8 exhibit 1H NMR doublets at 4.85 (Hα) and 2.38 (Hβ) ppm, with a three-bond coupling constant of 11.7 Hz. The corresponding 13C{1H} peaks appear at 71.6 (d, 2JCP = 4 Hz) and 44.4 (s) due to CHα and CHβ, respectively. The trimethylsilyl group is responsible for a singlet in the 1H NMR spectrum at 0.13 ppm and comparable data are found for the related trans isomer. Two-dimensional homonuclear and heteronuclear NMR experiments further support the formulations depicted in Scheme 8 for compounds 7 and 8.
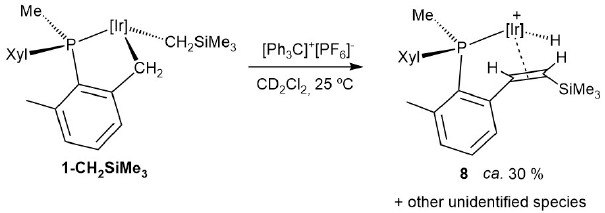
Scheme 8
Reaction of 1-CH2SiMe3with [Ph3C]+[PF6]-.
We performed an additional experiment to corroborate the proposed nature of compounds 7 and 8 and in support of our mechanistic suggestion of alkyl migratory insertion into an alkylidene functionality as the key C-C coupling step. Thus, we attempted the synthesis of the trimethylsilyl-substituted alkene complex 8 by α-hydride abstraction by Ph3C+ from alkyl compound 1-CH2 SiMe3 (Scheme 8),earlier reported by our group.[18b]The reaction proved to be more complex than foreseen and led to a mixture of compounds encompassing several iridium hydrides. Among the latter, the main species (ca. 30% overall yield by NMR) was, however, the expected complex 8 (Scheme 8), therefore corroborating the advanced hypothesis regarding the participation of reactive iridium alkylidenes in the formation of complexes 7 and 8 as in Scheme 7.
Conclusions
Five-membered iridacycles that contain the bulky phosphine PMe(Xyl)2 metalated at one of its benzylic γ-CH bonds and further stabilized by metal coordination to η5-C5Me5, readily generate uncommon cationic Ir(III) alkylidenes by electrophilic attack of Ph3C+. Hampering participation of the resulting iridium-carbene bond in migratory insertion reactions permitted isolation of cationic Ir(III) metal acyclicalkylidenes 3 and 4 of remarkable thermal stability, while on the contrary, the presence of an additional hydride or methyl ligand promoted further reactivity, in the latter case with formation of a new C-C bond. From these studies, which are summarized in Schemes 1 and 2, and from deuterium labeling experiments collected in Schemes 4-6, it can be concluded that Ph3C+ is capable to effect electrophilic attack at α and β positions of carbon-based ligands in our complex precursors, as well as at Ir-H sites.
Experimental Section
General considerations. All preparations and manipulations were carried out under oxygen free argon or nitrogen, using conventional Schlenk techniques and, when specified, at low temperature. Solvents were rigorously dried and degassed before use. Microanalyses were performed by the Microanalytical Service of the Instituto de Investigaciones Químicas (Sevilla, Spain). Infrared spectra were recorded on a Bruker Vector 22 spectrometer. Solution NMR spectra were recorded on Bruker Avance DPX-300, Avance DRX-400, Avance DRX-500, and 400 Ascend/R spectrometers. The 1H and 13C resonances of the solvent were used as the internal standard and the chemical shifts are reported relative to TMS, while 31P was referenced to external H3PO4. Allaromatic couplingsin the1H NMR spectra are of ca. 7.5 Hz. The crystal structures were determined in a Bruker-Nonius, X8Kappa diffractometer. 1-H, 1-Cl,1-Me and 1-CH2 SiMe3 were prepared according to literature methods.[18] Other chemicals were commercially available and used as received. CCDC nos. 1498592-1498595 contain the crystallographic data for compounds3, 4, k-5 and t-5. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Compound 1-CD3 . It was synthesized by a modified procedure from that employed for the preparation of 1-Me,[18] consisting in the addition of a diethyl ether solution of Mg(CD3)I (1M, 50 µL) to a CH2Cl2 solution of 1+ (50 mg, 0.035 mmol). The reaction mixture was stirred at room temperature for 15 min, then the volatiles were removed under vacuum and the residue extracted with pentane. The solvent was evaporated to dryness to give compound 1-CD3 as a pale yellow powder (13 mg, 62 %). Spectroscopic data matched those of 1-Me except for the 1H NMR resonance due to Ir-CH3 that was absent in the case of 1-CD3 .
Compound 3. A solid mixture of 1-Cl (50 mg, 0.08 mmol) and [Ph3C]+[PF6]- (32 mg, 0.08 mmol) was dissolved in CH2Cl2 (3 mL) at -20 ºC and stirred at room temperature for 5 minutes, resulting in a dark green solution. Addition of pentane (10 mL) led to the precipitation of a green solid which was washed with diethyl ether to yield complex 3 as a dark green powder (54 mg, 87 %). Suitable crystals for X-ray analysis were obtained by slow diffusion of pentane into a dichloromethane solution of the compound. Elemental analysis calcd. for C27H39ClF6IrP2: C, 42.27; H, 5.12;found: C, 42.3; H, 4.8.1H NMR(400 MHz, CD2Cl2, 25 ºC) δ: 16.61 (d, 1 H, 3JHP = 0.9 Hz, Ir=CH), 8.18 (d, 1 H, Ha), 7.87 (dd, 1 H, 4JHP = 3.4 Hz, Hc), 7.61 (td, 1 H, 5JHP = 2.4 Hz, Hb), 7.40 (td, 1 H, 5JHP = 2.1 Hz, He), 7.33, 7.06 (m, 1 H each, Hd, Hf), 2.56 (d, 3 H, 2JHP = 11.0 Hz, PMe), 2.55, 1.32 (s, 3 H each, Meβ, Meγ), 2.44 (s, 3 H, Meα), 1.66 (d, 15 H, 4JHP = 1.8 Hz, C5Me5). 13C NMR(100 MHz, CD2Cl2, 25 ºC) δ: 262.4 (1JCH = 153 Hz, Ir=CH), 165.2 (d, 2JCP = 29 Hz, C1), 145.6 (C3), 143.8 (d, 1JCP = 61 Hz, C2), 142.4 (d, 2JCP = 11 Hz, C4/6), 140.9 (d, 2JCP = 8 Hz, C4/6), 139.1 (d, 3JCP = 8 Hz, CHc), 135.2 (CHb), 132.5 (d, 4JCP = 2 Hz, CHe), 131.9, 131.0 (d, 3JCP = 10 Hz, CHd, CHf), 131.4 (d, 3JCP = 13 Hz, CHa), 121.0 (d, 1JCP = 54 Hz, C5), 108.7 (C5Me5), 25.6, 24.0 (d, 3JCP = 6 Hz, Meβ, Me γ), 20.4 (d, 3JCP = 2 Hz, Meα), 14.5 (d, 1JCP = 42 Hz, PMe), 8.8 (C5Me5).31P{1H} NMR (160 MHz, CD2Cl2, 25 ºC) δ: 20.8.
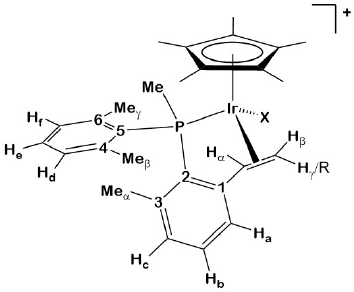
Fig. 7
Labeling scheme used for 1H and 13C{1H} NMR assignments.
Compound 4. A solution of [Ph3C]+[B(C6F5)4]- (40 mg, 0.04) in dichloromethane (0.5 mL) was added under argon over a solution of 2 (25 mg, 0.04) in the same solvent at -60 ºC. The solution turned dark-red immediately and, after addition of pentane, compound 4 precipitated as a red powder. Washing with pentane and drying under vacuum gave alkylidene 4 in 60% yield (31 mg). Crystals suitable for X-ray analysis were obtained by slow diffusion of pentane into a concentrated dichloromethane solution of the complex, however, due to its instability toward moisture and oxygen, reliable microanalytical data could not be obtained.1H NMR(400 MHz, CD2Cl2, 25 ºC) δ: 14.9 (s, 1 H, Ir=CH), 7.76 (d, 1 H, Ha), 7.55 (m, 2 H, Hc, He), 7.40, 7.25 (m, 1 H each, Hd, Hf), 7.35 (m, 1 H, Hb), 3.40 (d, 1 H, 2JHH = 8.6 Hz, IrCHH), 3.33 (d, 1 H, 2JHH = 8.6 Hz, IrCHH), 2.91, 2.16 (s, 3 H each, Meβ, Meγ), 2.27 (s, 3 H, Meα), 1.96 (d, 15 H, 4JHP = 1.8 Hz, C5Me5). 13C NMR (125 MHz, CD2Cl2, -60 ºC) δ: 243.7 (d, 2JCP = 28 Hz, Ir=CH), 162.4 (d, 2JCP = 27 Hz, C1), 144.8 (d, 2JCP = 4 Hz, C3), 143.9 (d, 2JCP = 16 Hz, C4/6), 141.5 (d, 2JCP = 6 Hz, C4/6), 134.4 (CHb, overlapped with BAr4-), 133.3 (d, 3JCP = 10 Hz, CHc), 129.8 (CHe), 129.1, 128.4 (d, 3JCP = 11 Hz, CHd, CHf), 127.2 (d, 3JCP = 17 Hz, CHa), 111.1 (d, 1JCP = 69 Hz, C5), 101.9 (C5Me5), 24.4, 19.2 (d, 3JCP = 6, 14 Hz, Meβ, Meγ), 19.0 (d, 3JCP = 5 Hz, Meα), 13.7 (d, 1JCP = 45 Hz, IrCH2), 8.5 (C5Me5). Signal corresponding to C2 was not identified.31P{1H} NMR (160 MHz, CD2Cl2, 25 ºC) δ: -13.1.
Reaction of 1-Me with [Ph3C]+ [B(C6 F5 )4 ]- (Compounds 5 and 6). A solution of [Ph3C]+[B(C6F5)4]- (92 mg, 0.10 mmol) in CH2Cl2 (1 mL) was added at 0 ºC under argon over a CH2Cl2 solution of 1-Me (60 mg, 0.10 mmol) placed in a Schlenk flask. The reaction mixture was stirred at room temperature for 30 min and analyzed by 31P{1H} NMR, which showed complete conversion of the methyl complex 1-Me into a ca.63:7:30 mixture of compounds k-5: t-5: 6. Heating the solution at 50 ºC for 12 hours resulted in complete isomerization of k-5 into t-5, while 6 remained unaltered. The solvent was removed under vacuum and the residue washed with diethyl ether. Compounds 5 and 6 were purified by crystallization from CH2Cl2/pentane to obtain a mixture of both isomers in 56 % overall yield (72 mg); however all attempts to isolate the individual complexes in a pure state were unsuccesful. Spectroscopic data obtained from mixtures of these isomers, are detailed below. Elemental analysis for the mixture of isomers calcd. for C52H42BF20IrP: C, 48.76; H, 3.31. Found: C, 48.9; H, 3.5.Compound t-5: 1H NMR (500 MHz, CD2Cl2, 25 ºC) δ: 7.47 (d, 1 H, Ha), 7.36 - 7.23 (m, 3 H, CHXyl), 7.07 - 6.98 (m, 2 H, CHXyl), 4.27 (dd, 1 H, 3JHH = 10.5, 3JHH = 8.6 Hz, Hα), 2.83 (dt, 1 H, 3JHH = 10.5, 2JHH = 3JHP = 2.4 Hz, Hγ), 2.64 (dd, 1 H, 3JHH = 8.6, 2JHH = 2.4 Hz, Hβ), 2.59, 1.45 (s, 3 H each, Meβ, Meγ), 2.46 (d, 3 H, 2JHP = 9.0 Hz, PMe), 1.91 (s, 3 H, Meα), 1.73 (d, 15 H, 4JHP = 1.7 Hz, C5Me5), -15.77 (d, 1 H, 2JHP = 30.1 Hz, IrH). 13C NMR (125 MHz, CD2Cl2, 25 ºC) δ: 146.9 (d, 2JCP = 23 Hz, C1), 141.8 (C3), 141.6, 140.2 (d, 2JCP = 9 Hz, C4, C6), 136.8 (d, 1JCP = 61 Hz, C2), 131.8, 131.5 (CHb, CHe), 131.1, 130.3 (d, 3JCP = 8 Hz, CHd, CHf), 130.7 (d, 3JCP = 8 Hz, CHc), 127.0 (d, 3JCP = 16 Hz, CHa), 123.4 (d, 1JCP = 52 Hz, C5), 101.0 (C5Me5), 62.7 (d, 2JCP = 4 Hz, CHα), 34.1 (CHβHγ), 28.0 (d, 1JCP = 50 Hz, PMe), 24.7, 22.8 (d, 3JCP = 7 Hz, Meβ, Me γ), 19.9 (d, 3JCP = 18 Hz, Meα), 8.4 (C5Me5).31P{1H} NMR(200 MHz, CD2Cl2, 25 ºC) δ: -0.1.Compound k-5:1H NMR(500 MHz, CD2Cl2, 25 ºC) δ: 7.40 (m, 1 H, Ha), 7.35 - 7.19 (m, 3 H, CHXyl), 7.08 - 6.96 (m, 2 H, CHXyl), 4.41 (t, 1 H, 3JHH, 3JHH = 9.1 Hz, Hα), 2.67 (m, overlapped, Hγ), 2.65, 1.72 (s, 3 H each, Meβ, Meβ), 2.24 (d, 3 H, 2JHP = 10.2 Hz, PMe), 2.12 (t, 1 H, 3JHH = 2JHH = 9,1 Hz, Hγ), 1.93 (d, 15 H, 4JHP = 1.8 Hz, C5Me5), 1.89 (s, 3 H, Meα), -14.31 (d, 1 H, 2JHP = 28.5 Hz, IrH).31P{1H} NMR(200 MHz, CD2Cl2, 25 ºC) δ: -9.8.Compound 6: 1H NMR(500 MHz, CD2Cl2, 25 ºC) δ: 7.36 - 7.23 (m, 4 H, CHXyl), 7.07 - 6.98 (m, 2 H, CHXyl), 5.30 (d, 1 H, 4JHP = 4.2 Hz, Hα/β), 4.54 (s, 1 H, Hα/β), 3.71 (d, 1 H, 2JHH = 13.9 Hz, IrCHH), 2.99 (d, 1 H, 2JHH = 13.9 Hz, IrCHH), 2.69, 1.45 (s, 3 H each, Meβ, Meγ), 2.43 (d, 3 H, 2JHP = 9.6 Hz, PMe), 2.01 (s, 3 H, Meα), 1.80, 1.75 (d, 3 H each, 4JHP = 2.8 Hz, 2 Cp-Me), 1.57 (s, 3 H, Cp-Me), 0.85 (d, 3 H, 3JHP = 2.7 Hz, IrCH3), 0.76 (d, 3 H, 4JHP = 1.4 Hz, Cp-Me).13C NMR(125 MHz, CD2Cl2, 25 ºC) δ: 152.7 (d, 2JCP = 27 Hz, C1), 141.5, 140.1 (C4/6), 140.2 (C3), 134.8 (d, 1JCP = 65 Hz, C2),129.4 (d, 3JCP = 9 Hz, CHc), 132.1 (CHb, CHe), 131.2, 131.0 (d, 3JCP = 9 Hz, CHd, CHf), 127.5 (d, 3JCP = 16 Hz, CHa), 124.8 (d, 1JCP = 53 Hz, C5), 114.2 111.8, 110.1, 109.4, 109.3 (C5Me4CHαHβ), 68.6 (CHαHβ), 29.6 (IrCH2), 25.3, 24.7 (d, 3JCP = 6 Hz, Meβ, Me γ), 21.0 (d, 1JCP = 44 Hz, PMe), 20.1 (d, 3JCP = 16 Hz, Meα), 8.2, 7.8, 6.8, 4.7 (C5Me4CHαHβ), -10.9 (d, 2JCP = 3 Hz, IrCH3).31P{1H} NMR (200 MHz, CD2Cl2, 25 ºC) δ: 12.2.
Compound 7. To a solid mixture of 1-Cl (50 mg, 0.08 mmol) and NaBArF (72 mg, 0.08 mmol) placed in a Schlenk flask were added 5 mL of CH2Cl2. The reaction mixture was stirred for 5 min at room temperature and ethyldiazoacetate (10 µL, 0.096 mmol) was added. The solution was filtered and the volatiles were evaporated under reduced pressure to obtain 7 (102 mg, 86 %) as a pale yellow powder mixture of isomers in a ratio ofca. 60:40. Tentative isomer assignement for each set of signals is based on 2D-NOESY analysis, since NOE cross-peaks can be found between Hα and Hβ only for the cis isomer, whereas the trans isomer presents a NOE cross-peak between Hα and Ha, which cannot be detected in the former case.The two isomers were jointly recrystallized from a 1:2 mixture of CH2Cl2: pentane. Elemental analysis for the mixture of isomerscalcd.for C63H53BF24IrO2P: C, 49.39; H, 3.49,found: C, 49.0; H, 3.0.Trans-7 (60%): IR (Nujol): υ(IrH) 2175, υ(CO) 1710 cm-1.1H NMR(400 MHz, CD2Cl2, 25 ºC) δ: 7.57 (d, 1 H, Ha), 7.37, 7.30, 7.08 (m, Hb-f, overlapped with an analogous signals of the minor isomer), 4.73 (d, 1 H, 3JHH = 9.3 Hz, CHα), 4.29 (m, 1 H, OCHHCH3), 4.14 (m, 1 H, OCHHCH3), 3.72 (d, 1 H, 3JHH = 9.3 Hz, CHβ), 2.61, 1.46 (s, 3 H each, Meβ, Meγ), 2.56 (d, 3 H, 2JHP = 10.0 Hz, PMe), 1.95 (s, 3 H, Meα), 1.73 (d, 15 H, 4JHP = 1.7 Hz, C5Me5), 1.32 (t, 3 H, 3JHH = 7.1 Hz, OCH2CH3), -15.35 (d, 1 H, 2JHP = 27.8 Hz, IrH).13C NMR(100 MHz, CD2Cl2, 25 ºC) δ: 170.0 (CO), 147.6 (d, 2JCP = 26 Hz, C1), 142.3, 141.7, 141.5, 141.1 (C2, C3, C4, C6), 132.7 (CHb), 132.2 (d, 4JCP = 2 Hz, CHe), 131.5 (CHd/f), 131.4 (CHc), 131.0 (d, 3JCP = 6 Hz, CHd/f), 127.1 (d, 3JCP = 16 Hz, CHa), 122.7 (d, 1JCP = 56 Hz, C5), 103.1 (C5Me5), 61.7 (OCH2CH3), 59.5 (d, 2JCP = 4 Hz, CHα), 40.9 (CHβ),26.5 (d, 1JCP = 47 Hz, PMe), 25.0, 23.7 (d, 3JCP = 6 Hz, Meβ, Meγ), 20.3 (d, 3JCP = 6 Hz, Meα), 14.5 (OCH2CH3), 8.8 (C5Me5).31P{1H} NMR(160 MHz, CD2Cl2, 25 ºC) δ: 1.0.Cis-7(40 %):IR(Nujol): υ(IrH) 2150, υ(CO) 1650 cm-1.1H NMR(400 MHz, CD2Cl2, 25 ºC) δ: 7.46 (d, 1 H, Ha), 7.37, 7.30, 7.08 (m, Hb-f, overlapped with analogous signals of the major isomer), 4.67 (d, 1 H, 3JHH = 9.1 Hz, CHα), 4.02 (m, 1 H, OCHHCH3), 3.96 (m, 1 H, OCHHCH3), 3.49 (d, 1 H, 3JHH = 9.1 Hz, CHβ), 2.61, 1.53 (s, 3 H each, Meβ, Meγ), 2.50 (d, 3 H, 2JHP = 11.1 Hz, PMe), 1.92 (s, 3 H, Meα), 1.75 (d, 15 H, 4JHP = 1.6 Hz, C5Me5), 1.16 (t, 3 H, 3JHH = 7.1 Hz, OCH2CH3), -16.39 (d, 1 H, 2JHP = 27.8 Hz, IrH).13C NMR (100 MHz, CD2Cl2, 25 ºC) δ: 172.5 (CO), 146.4 (d, 2JCP = 23 Hz, C1), 142.2, 141.5, 141.4, 141.0 (C2, C3, C4, C6), 132.4 (d, 4JCP = 2 Hz, CHe), 132.0 (CHb), 131.6 (CHd/f), 131.3 (CHc), 130.9 (d, 3JCP = 6 Hz, CHd/f), 126.8 (d, 3JCP = 17 Hz, CHa), 124.7 (C5), 103.1 (C5Me5), 63.9 (d, 2JCP = 3 Hz, CHα), 61.4 (OCH2CH3), 40.3 (CHβ), 25.7 (d, 1JCP = 47 Hz, PMe), 25.6, 23.4 (d, 3JCP = 5 Hz, 8 Hz, Meβ, Meγ), 20.1 (d, 3JCP = 6 Hz, Meα), 14.0 (OCH2CH3), 8.8 (C5Me5).31P{1H} NMR(160 MHz, CD2Cl2, 25 ºC) δ: 3.1.
Compound 8. To a solid mixture of 1-Cl (50 mg, 0.081 mmol) and NaBArF (72 mg, 0.081 mmol) placed in a Schlenk flask 3 mL of CH2Cl2 were added at -20 ºC. After stirring for 10 min, a solution of N2CHSiMe3 (2M in hexanes, 50 µL) was added at this temperature and the reaction allowed to warm up to 25 ºC and additionally stirred for 1 hour. The solution was filtered and the volatiles were removed under vacuum to give a pale yellow foam which was washed with pentane. 1H and 31P{1H} NMR spectroscopic analysis of the crude showed four different products in a ratio of ca. 53:40:4:3, all of which exhibited characteristic Ir-H resonances in the 1H NMR spectrum. The two major species were identified as the trans (53 %) and cis (40 %) isomers. However, only cis-8 was isolated as a single species by crystallization from Et2O/pentane (1:1) in 30 % yield (38 mg).Cis-8 (40 %):IR(Nujol): υ(IrH) 2160 cm-1. Elemental analysis calcd.for C63H57BF24IrPSi: C, 49.39; H, 3.75,found: C, 49.3; H, 3.6.1H NMR (500 MHz, CD2Cl2, 25 ºC) δ: 7.41 (d, 1 H, Ha), 7.34 (m, 2 H, Hb, He), 7.26, 7.01 (dd, 1 H each, 4JHP = 4.2 Hz, Hd, Hf), 7.05 (dd, 1 H, 4JHP = 4.1 Hz, Hc), 4.85 (d, 1 H, 3JHH = 11.7 Hz, CHα), 2.62, 1.53 (s, 3 H each, Meβ, Meγ), 2.52 (d, 3 H, 2JHP = 10.5 Hz, PMe), 2.38 (d, 1 H, 3JHH = 11.7 Hz, CHβ), 1.89 (s, 3 H, Meα), 1.70 (d, 15 H, 4JHP = 1.6 Hz, C5Me5), 0.13 (s, 9 H, SiMe3), -15.85 (d, 1 H, 2JHP = 30.7 Hz, IrH).13C NMR(125 MHz, CD2Cl2, 25 ºC) δ: 147.2 (d, 2JCP = 27 Hz, C1), 142.2, 140.5 (d, 2JCP = 10, 8 Hz, C4, C6), 141.6 (C3), 132.3, 131.6 (CHb, CHe), 131.3 (d, 3JCP = 10 Hz, CHd/f), 130.9 (m, CHc, CHd/f), 128.8 (C2, overlapped with BArF), 127.3 (d, 3JCP = 17 Hz, CHa), 124.4 (d, 1JCP = 49 Hz, C5), 101.0 (C5Me5), 71.6 (d, 2JCP = 4 Hz, CHα), 44.4 (CHβ),28.4 (d, 1JCP = 48 Hz, PMe), 25.8, 22.5 (d, 3JCP = 5, 8 Hz, Meβ, Meγ), 19,8 (d, 3JCP = 4 Hz, Meα), 8.6 (C5Me5), 0.0 (SiMe3).31P{1H} NMR(200 MHz, CD2Cl2, 25 ºC) δ: -7.6. Trans-8 (53%): It could not be isolated in pure form, but representative spectroscopic data include: 1H NMR (500 MHz, CD2Cl2, 25 ºC)4.36 (d, 3JHH = 12.7 Hz, CHα), 2.27 (PMe and CHβ), 1.94 (C5Me5), 0.26 (SiMe3) and -16.84 (d, 2JCP = 30.7 Hz, Ir-H) ppm. 31P{1H} NMR(200 MHz, CD2Cl2, 25 ºC) δ: 0.9 ppm.
Acknowledgments
Financial support from Spanish Ministry of Science (Projects CTQ2013-42501-P, CTQ2014-51912-REDC and CTQ-2014-52769-C3-3-R) and the Junta de Andalucía (Grant FQM-119 and project P09-FQM-4832 and FQM-2126) is gratefully acknowledged. J.C. thanks the EU 7th Framework Program, Marie Skłodowska-Curie actions (COFUND, Grant Agreement no. 267226) and Junta de Andalucía for a Talentia Postdoc Fellowship.
References
1.. 1. (a) Arduengo, J. A.; Bertrand, G. Chem. Rev. 2009, 109, 3209-3884 (special issue dedicated to carbenes). (b) Crabtree, R. H. J. Organomet. Chem. 2005, 690, 5451-5457; (c) Díez-González, S., Nolan, S.P. Coord. Chem. Rev. 2007, 251, 874-883.
2.. 2. (a) Lavallo, V.; Canac, Y.; Prasang, C.; Donnadieu, B.; Bertrand, G. Angew. Chem. Int. Ed. 2005, 44, 5705-5709; (b) Jazzar, R.; Dewhurst, R. D.; Bourg, J. B.; Donnadieu, B.;Canac, Y.; Bertrand, G. Angew. Chem. Int. Ed. 2007, 46, 2899-2902; (c) Zeng, X.; Frey, D. G.; Kinjo, R.; Donnadieu, B.; Bertrand, G. J. Am. Chem. Soc. 2009, 131 ,8690-8696; (d) Chu, J., Munz, D., Jazzar, R., Melaimi, M., Bertrand, G. J. Am. Chem. Soc. 2016, 138, 7884-7887; (e) Roy, S.; Mondal, K. C.;Roesky, H. W. Acc. Chem. Res. 2016, 49, 357-369.
3.. 3. Selected examples: (a) Miki, S.; Ogno, T.; Iwasaki, H.; Yoshida, Z.-I. J. Phys. Org. Chem. 1988, 1, 333-338; (b) Lu, Z.; Jones, W. M.; Winchester, W. R. Organometallics 1993, 12, 1344-1350; (c) Öfele, K.; Tosh, E.; Taubmann, C.; Herrmann, W. A. Chem. Rev. 2009, 109, 3408-3444.
4.. 4. (a) Carmona, E.; Paneque, M.; Poveda, M. L. Dalton Trans. 2003, 4022-4029; (b) Conejero, S.; Paneque, M.; Poveda, M. L.; Santos, L. L.; Carmona, E. Acc. Chem. Res. 2010, 43, 572-580; (c) Werner, H. Angew. Chem., Int. Ed. 2010, 49, 4714; (d) Whited, M. T.; Grubbs, R. H. Acc. Chem. Res. 2009, 42, 1607-1616; (e) Valpuesta, J. E. V.; Álvarez, E.; López-Serrano, J.; Maya, C.; Carmona, E. Chem. Eur. J. 2009, 18, 13149-13159.
5.. 5. (a) Hoover, J. F.; Stryker, J. M. J. Am. Chem. Soc. 1990, 112, 464-465; (b) Casty, G. L.; Stryker, J. M. Organometallics 1997, 16, 3083-3085; (c) Holtcamp, M. W.; Henling, L. M.; Day, M. W.; Labinger, J. A.; Bercaw, J. E. Inorg. Chim. Acta 1998, 270, 467-478; (d) Owen, J. S.; Labinger, J. A.; Bercaw, J. E. J. Am. Chem. Soc. 2006, 128, 2005-2016; (e) Lee, D.-H-; Chen, J.; Faller, J. W.; Crabtree, R. Chem. Commun. 2001, 213-214; (f) Clot, E.; Chen, J.; Lee, D.-H.; Sung, S. Y.; Appelhans, L. N.; Faller, J. W.; Crabtree, R. H.; Eisenstein, O. J. Am. Chem. Soc. 2004, 126, 8795-8804.
6.. 6. (a) Herndon, J. W. Coord. Chem. Rev. 2011, 255, 3 (annual survey covering the year 2009; see references therein for previous years); (b) Caulton, K. G. J. Organomet. Chem. 2001, 56, 617-618.
7.. 7. (a) Dötz, K. H. Metal Carbenes in Organic Synthesis. Springer, 2004; (b) Dörwald, F. Z. Metal Carbenes in Organic Synthesis. Wiley-VCH: Weinheim, Germany, 2008; (c) Moss, R. A.; Doyle, M. P. Contemporary Carbene Chemistry. John Wiley & Sons, 2014.
8.. 8. For early review articles see: (a) Aguero, A.; Osborn, J. A. New J. Chem. 1988, 12, 111-118; (b) Brothers, P. J.; Roper, W. Chem. Rev. 1988, 88, 1293-1326; (c) Gallop, M. A.; Roper, W. R. Adv. Organomet. Chem. 1986, 25, 121-198; (d) Herrmann, W. A. Adv. Organomet. Chem. 1982, 20, 159-263.
9.. 9. (a) Chauvin, Y. Angew. Chem. Int. Ed. 2006, 45, 3740-3747; (b) Schrock, R. R. Angew. Chem. Int. Ed. 2006, 45, 3748-3759; (c) Grubbs, R. H. Angew. Chem. Int. Ed. 2006, 45, 3760-3765.
10.. 10. For recent examples see: (a) Poverenov, E.; Milstein, D. Chem. Commun. 2007, 3189-3191; (b) Burford, R. J.; Piers, W. E.; Parvez, M. Organometallics 2012, 31, 2949-2952; (c) Doyle, L. E.; Piers, W. E.; Borau-Garcia, J.; Sgro, M. J.; Spasyuka, D. M. Chem. Sci. 2016, 7, 921-931; (d) Yuan, J.; Hughes, R. P.; Rheingold, A. L. Dalton Trans. 2015, 44, 19518-19527.
11.. 11. (b) Fryzuk, M. D.; Macneil, P. A.; Rettig, S. J. J. Am. Chem. Soc. 1985, 107, 6708-6710. (c) Fryzuk, M. D.; Gao, X.; Joshi, K.; MacNeil, P. A.; Massey, R. L. J. Am. Chem. Soc. 1993, 115, 10581-10590; (d) Klein, D. P.; Bergman, R. G. J. Am. Chem. Soc. 1989, 111, 3079-3080; (e) Campos, J.; Peloso, R.; Brookhart, M.; Carmona, E. Organometallics2013, 32, 3423-3426.
12.. 12. (a) Lara, P.; Paneque, M.; Poveda, M. L.; Santos, L. L.; Valpuesta, J. E. V.; Carmona, E.; Moncho, S.; Ujaque, G.; Lledós, A.; Álvarez, E.; Mereiter, K. Chem. Eur. J. 2009, 15, 9034-9045; (b) Lara, P.; Paneque, M.; Poveda, M. L.; Santos, L. L.; Valpuesta, J. E. V.; Salazar, V.; Carmona, E.; Moncho, S.; Ujaque, G.; Lledós, A.; Maya, C.; Mereiter, K. Chem. Eur. J. 2009, 15, 9046-9057; (c) Paneque, M.; Poveda, M. L.; Santos, L. L.; Carmona, E.; Lledós, A.; Ujaque, G.; Mereiter, K. Angew. Chem. Int. Ed. 2004, 43, 3708-3711.
13.. 13. (a) Alías, F. M.; Poveda, M. L.; Sellin, M.; Carmona, E. J. Am. Chem. Soc. 1998, 120, 5816-5817; (b) Besora, M.; Vyboishchikov, S. F.; Lledós, A.; Maseras, F.; Carmona, E.; Poveda, M. L. Organometallics 2010, 29, 2040-2045; (c) Alías, F. M.; Poveda, M. L.; Sellin, M.; Carmona, E.; Gutiérrez, E.; Monge, A. Organometallics 1998, 17, 4124-4126.
14.. 14. (a) Paneque, M.; Posadas, C. M.; Poveda, M. L.; Rendon, N.; Mereiter, K. Organometallics 2007, 26, 1900-1906; (b) Luecke, H. F.; Bergman, R. G. J. Am. Chem. Soc. 1998, 120, 11008-11009; (d) Thorn, D. L.; Tulip, T. H. J. Am. Chem. Soc. 1981, 103, 5984-5986; (e) Thorn, D. L. Organometallics 1982, 1, 879-885; (f) Thorn, D. L.; Tulip, T. H. Organometallics 1982, 1, 1580-1586; (g) Bell, T. W.; Haddleton, D. M.; McCamley, A.; Partridge, M. G.; Perutz, R. N.; Willner, H. J. Am. Chem. Soc. 1990, 112, 9212-9226; (h) France, M. B.; Feldman, J.; Grubbs, R. H. J. Chem. Soc., Chem. Commun. 1994, 1307-1308; (i) Bleeke, J. R.; Behm, R. J. Am. Chem. Soc. 1997, 119, 8503-8511.
15.. 15. Choi, J.; Wang, D. Y.; Kundu, S.; Choliy, Y.; Emge, T. J.; Krogh-Jespersen, K.; Goldman, A. S. Science 2011, 332, 1545-1548.
16.. 16. Polukeev, A. V.; Marcos, R.; Ahlquistb, M. S. G.; Wendt, O. F. Chem. Sci. 2015, 6, 2060.
17.. 17. (a) Arndtsen, B. A.; Bergman, R. G. Science 1995, 270, 1970; (b) Burger, P.; Bergman, R. G. J. Am. Chem. Soc. 1993, 115, 10462.
18.. 18. (a) Campos, J.; Esqueda, A. C.; Carmona, E. Chem. Eur. J. 2010, 16, 419-422; (b) Campos, J.; Álvarez, E.; Carmona, E. New J. Chem. 2011, 35, 2122-2129; (c) Rubio, M.; Campos, J.; Carmona, E.Org. Lett. 2011, 13, 5236 -5239;(d) Campos, J.; Rubio, M.; Esqueda, A. C.; Carmona, E. J. Label. Compd. Radiopharm. 2012, 55, 29-38.
19.. 19. Campos, J.; Carmona, E. Organometallics 2015, 34, 2212-2221.
20.. 20. (a) Campos, J.; Esqueda, A. C.; López-Serrano, J.; Sánchez, L.; Cossio, F. P.; de Cozar, A.; Álvarez, E.; Maya, C.; Carmona, E. J. Am. Chem. Soc. 2010, 132, 16765-16767; (b) Campos, J.; López-Serrano, J.; Álvarez, E.; Carmona, E. J. Am. Chem. Soc. 2012, 134, 7165-7175; (c) Campos, J.; Espada, M. F.; López-Serrano, J.; Carmona, E. Inorg. Chem. 2013, 52, 6694-6704.
21.. 21. Espada, M. F.; López-Serrano, J.; Poveda, M. L.; Carmona, E. Angew. Chem. Int. Ed. 2015, 54, 8751-8755.
22.. 22. Crabtree R. H. The Organometallic Chemistry of the Transition Metals. Wiley, 2014.
23.. 23. Hartwig, J. F. Organotransition Metal Chemistry: From Bonding to Catalysis. University Science Books. 2010.
24.. 24. (a) Hayes, J. C.; Cooper, N. J. J. Am. Chem. Soc. 1982, 104, 5570-557; (b) Guerchais, V.; Lapinte, C. J. Chem. Soc. Chem. Commun. 1986, 663-664.
25.. 25. (a) Campos, J.; Peloso, R.; Carmona, E. Angew. Chem. Int. Ed. 2012, 51, 8255 - 8258; (b) Campos, J.; Ortega-Moreno, L.; Conejero, S.; Peloso, R.; López-Serrano, J.; Maya, C.; Carmona, E., Chem. Eur. J. 2015, 21, 8883-8896.
26.. 26. Liu, L.; Zhang, J. Chem. Soc. Rev. 2016, 45, 506-516.
27.. 27. See for example: Cheng, T.-Y.; Brunschwig, B. S.; Bullock, R. M. J. Am. Chem. Soc. 1998, 120, 13121-13137.
28.. 28. Sattler, A.; Parkin, G. Chem. Commun. 2011, 47, 12828-12830.
29.. 29. (a) Hinderling, C.; Feichtinger, D.; Plattner, D.; Chen, P. J. Am. Chem. Soc. 1997, 119, 10793-10804; (b) Shinomoto, R. S.; Desrosiers, P. J.; Harper, T. G. P.; Flood, T. C. J. Am. Chem. Soc. 1990, 112, 704-713; (c) Casalnuovo, A. L.; Calabrese, J. C.; Milstein, D. J. Am. Chem. Soc. 1988, 110, 6738-6744; (d) Wenzel, T. T.; Bergman, R. G. J. Am. Chem. Soc. 1986, 108, 4856-4867.
30.. 30. See for instance: (a) Klahn, A. H.; Oelckers, B.; Godoy, F.; Garland, M. T.; Vega, A.; Perutz, R. N.; Higgitt, C. L. J. Chem. Soc., Dalton Trans. 1998, 3079-3086; (b) Fairchild, R. M.; Holman, K. T. Organometallics 2008, 27, 1823; (c) Fan, L.; Turner, M. L.; Hursthouse, M. B.; Malik, K. M. A.; Gusev, O. V.; Maitlis, P. M. J. Am. Chem. Soc. 1994, 116, 385; (d) Bandy, J. A.; Mtetwa, V. S. B.; Prout, K.; Green, J. C.; Davies, C. E.; Green, M. L. H.; Hazel, N. J.; Izquierdo, A.; Martin-Polo, J. J. J. Chem. Soc., Dalton Trans. 1985, 2037-2049.
31.. 31. (a) Alías, F. M.; Daff, P. J.; Paneque, M.; Poveda, M. L.; Carmona, E.; Pérez, P. J.; Salazar, V.; Alvarado, Y.; Atencio, R.; Sánchez-Delgado, R. Chem. Eur. J. 2002, 8, 5132-5146; (b) Paneque, M.; Poveda, M. L.; Santos, L. L.; Carmona, E.; Lledós, A.; Ujaque, G.; Mereiter, K. Angew. Chem. Int. Ed. 2004, 43, 3708-3711; (c) Lara, P.; Paneque, M.; Poveda, M. L.; Santos, L. L.; Valpuesta, J. E. V.; Carmona, E.; Moncho, S.; Ujaque, G.; Lledós, A.; Álvarez, E.; Mereiter, K. Chem. Eur. J. 2009, 15, 9034-9045; (d) Lara, P.; Paneque, M.; Poveda, M. L.; Santos, L. L.; Valpuesta, J. E. V.; Salazar, V.; Carmona, E.; Moncho, S.; Ujaque, G.; Lledós, A.; Maya, C.; Mereiter, K. Chem. Eur. J. 2009, 15, 9046-9057.
32.. 32. Jellema, E.; Jongerius, A. L.; Reek, J. N. H.; de Bruin, B. Chem. Soc. Rev. 2010, 39, 1706.
33.. 33. See for example: (a) Caballero, A.; Despagnet-Ayoub, E.; Díaz-Requejo, M. M.; Díaz-Rodríguez, A.; González-Núñez, M. E.; Mello, R.; Muñoz, B. K.; Ojo, W.-S.; Asensio, G.; Etienne, M.; Pérez, P. J. Science 2011, 332, 835-838; (b) Zheng, J.; Lin, J.-H.; Yu, L.-Y.; Wei, Y.; Zheng, X.; Xiao, J.-C. Org. Lett. 2015, 17, 6150-6153; (c) Holzwarth, M. S.; Alt, I; Plietker, B. Angew. Chem. Int. Ed. 2012, 51, 5351-5354; (d) Muller, P. Acc. Chem. Res. 2004, 37, 243-251.
34.. 34. (a) Braga, A. A. C.; Caballero, A.; Urbano, J.; Díaz-Requejo, M. M.; Pérez, P. J.; Maseras, F. ChemCatChem 2011, 3, 1646. (b) Hansen, J. H.; Parr, B. T.; Pelphrey, P.; Jin, Q.; Autschbach, J.; Davies, H. M. L. Angew. Chem. Int. Ed. 2011, 50, 2544.
Notes
Associated Content Supporting Information: kinetic studies on the isomerization of compound 5 and X-ray crystallographic studies.
Author notes
In memory of Professor Roberto Sánchez Delgado, dear friend and outstanding leader and scientist.
Avenida Américo Vespucio 49, 41092 Sevilla (Spain). Fax: (+34) 954460565 E-mail: guzman@us.es
