Journal of the Mexican Chemical Society, vol. 61, no. 4, 2017
Sociedad Química de México A.C.
Jun Hu
Anhui University, China
Wei Ma
Anhui Medical University, China
Ning Li *
Anhui Medical University, China
Kai-Jin Wang *
Anhui University, China
Date received: 05 April 2017
Date accepted: 23 June 2017
Funding
Funding source: Zhenxing Plan of Education Department of Anhui Province
Contract number: KJ2016SD03
Funding
Funding source: Anhui University
Contract number: Y01008410
Abstract: Chuju, the flower of Chrysanthemum morifolim cv. Chuju, is a traditional medicine-food material used in China. Bioassay-guided chemical investigation on Chuju led the isolation of 16 flavonoids (1-16) characterized as quercetin (1), isorhamnetin 3-O-b-D-glucoside (2), eriodictyol (3), pyracanthoside (4), apigenin (5), apigetrin (6), acacetin (7), acacipetalin (8), luteolin (9), diosmetin (10), spinacetin (11), axillarin (12), bonanzin (13), cirsiliol (14), chrysosplenol D (15) and artemetin (16). Among them, compounds (11-16) were first reported from the Chrsanthemum species. Chuju flavonoids displayed strong antioxidant in DPPH (1, 1-diphenyl-2- picrylhydrazyl) radical scavenging assay, and high anti-inflammatory activity by inhibiting NO production in LPS-induced RAW 264.7 cells. These findings suggests that Chuju is an ideal source of natural flavonoids with significant antioxidant and anti-inflammatory effects, and is worthy of further development as health-promoting food or supplement.
Keywords: Chrysanthemum morifolim cv. Chuju, Chuju flowers, flavonoids, antioxidant activity, anti-inflammatory activity.
Resumen: La flor de Chrysanthemum morifolim cv. Chuju, conocida popularmente como Chuju, se utiliza tradicionalmente como alimento y medicina en China. Un estudio químico biodirigido de un extracto de las flores condujo al aislamiento de 16 flavonoides, los cuales se caracterizaron como quercetina (1), isoramnetina 3-O-b-D-glucosido (2), eriodictiol (3), piracantosido (4), apigenina (5), apigetrina (6), acacetina (7), acacipetalina (8), luteolina (9), diosmetina (10), espinacetina (11), axilarina (12), bonanzina (13), cirsiliol (14), crisosplenol D (15) and artemetina (16). Los productos 11-16 se describen por primera vez en la especie. Los flavonoides de Chuju demostraron una notable actividad antioxidante en el ensayo de captación del radical 2,2- difenil-1-picrilhidrazilo (DPPH); asimismo presentaron un fuerte efecto antiinflamatorio in vitro, valorado a través de la inhibición de la producción de NO en macrófagos RAW 264.7 estimulados con LPS. Estos resultados indican que Chuju es una fuente importante de flavonoides antiinflamatorios y antioxidantes con un potencial importante para su desarrollo como agente nutracéutico.
Palabras clave: Chrysanthemum morifolim cv. Chuju, flores de Chuju, flavonoids, actividad antioxidante, actividad antiinflamatoria.
Introduction
The oxidative stress emerges from the excessive generation of reactive oxygen species (ROS). Persistent oxidative stress may cause severe damages to proteins, lipids, DNA, and even result in cell death 1. These damages are often involved in the activation of chronic inflammatory pathways leading to the progression of diseases including cancer, neurodegenerative disorders, atherosclerosis, cardiovascular diseases, diabetes, chronic kidney diseases, and aging processes 2. Antioxidants are considered beneficial for biological systems to scavenge ROS and protect cells from oxidative and inflammatory damages 3. Synthetic antioxidants, such as propyl gallate (PG), butylated hydroxyanisole (BHA), tertiary butyhydroquinone (TBHQ) and butylated hydroxytoluene (BHT), are widely used as food additives 4. However, demands for naturally occurring antioxidants have been increasing due to the concerns about the safety of synthetic antioxidants 5. As a class of natural antioxidants, flavonoids have aroused much attention, and a lot of efforts have been made to search for antioxidant agents from plants rich in flavonoids 6.
Chrysanthemi Flos (CFs), termed as “Juhua” in Chinese, is the dried flower of Chrysanthemum morifolium Ramat (Asteraceae), which is a famous herbal medicine and food material in Southeast Asian countries such as China, South Korea, Japan and Singapore. C. morifolium was first originated in China, and has been cultivated for over 2000 years, forming a large number of medicinal and horticultural cultivars during the long evolution. Hangju (C. morifolium cv. Hangju), Chuju (C. morifolium cv. Chuju), Gongju (C. morifolium cv. Gongju) and Boju (C. morifolium cv. Boju) are the four famous medicinal cultivars of C. morifolium recorded in Chinese Pharmacopeia. Chuju originated in Chuzhou, Anhui Province, China. Chuju was once ranked first among the four famous medicinal cultivars of C. morifolium and chosen as the royal tribute in Qing Dynasty, and honored the national protected geographical indication (PGI) in 2002. Chuju was recorded the functions of scattering cold, clearing away heat and toxins, and brightening eyes, and traditionally used for the treatment of head dizziness, pain and swelling of eyes, particular for diseases caused by upward-disturbance of liver-yang 7.
Previous studies demonstrated that CFs possessed anti-oxidant, cardiovascular protection 8, anti-inflammation 9, hepato-protection 10, and anticancer 11 effects. Essential oil 12,13, terpenoids 9, flavonoids 14 and caffeoylquinic acids 15 in CFs have been characterized. However, almost all the previous reports related to CFs were focused on Hangju, and no detailed investigation on Chuju has been carried out till now.
Our preliminary experiments showed that Chuju were rich in flavonoids. On the other hand, we found that the EtOH extract exhibited considerable antioxidant activity in 1, 1-diphenyl-2-picrylhydrazyl radical (DPPH) assay and strongly inhibited the NO production in lipopolysaccharide (LPS)-induced murine macrophage RAW 264.7 cells. These results promoted us to perform a detailed bioassay guided chemical investigation on Chuju, which led the isolation of 16 compounds from two flavonoids-enriched fractions. Herin, the isolation, structural determination, antioxidant and anti-inflammatory activities of these flavonoids were described.
Results and Discussion
Bioassay guided isolation and structural identification of flavonoids 1-16
The 85% ethanol extract of the flowers of Chuju was partitioned by D101 macroporous resin CC to give four fractions (A-D). In DPPH radical-scavenging assay, the antioxidant activity order of the four fractions was C > B > D > A; the same order was shown in the experiment regarding the inhibitory activity on NO production in LPS-induced RAW 264.7 cells (Table 1). In total flavonoids content (TFC) experiment, fractions C showed the highest level of TFC value, flowed by D, B and A (Table 1). Thus, further purification was focused on the two flavonoids-enriched fractions (C and D). The isolated compounds (Fig.1) included quercetin (1) 16, isorhamnetin 3-O-b-D-glucoside (2) 17, eriodictyol (3) 18, pyracanthoside (4) 19, apigenin (5) 20, apigetrin (6) 20, acacetin (7) 21, acacipetalin (8) 21, luteolin (9) 16, diosmetin (10) 22, spinacetin (11) 16, axillarin (12) 16, bonanzin (13) 23, cirsiliol (14) 24, chrysosplenol D (15) 25 and artemetin (16) 23, and were characterized by detailed spectroscopic analysis and comparing with the previous literature data. Among them, compounds 11-16 have never been reported from Chrsanthemum species before, and 14 compounds (1-3, 5-7, 9-16) were isolated from Chuju for the first time.
Total flavonoid content (TFC), DPPH radical-scavenging activities and NO production inhibitory activities of the ethanol extract of the flower of Chuju and its fractions (A-D)
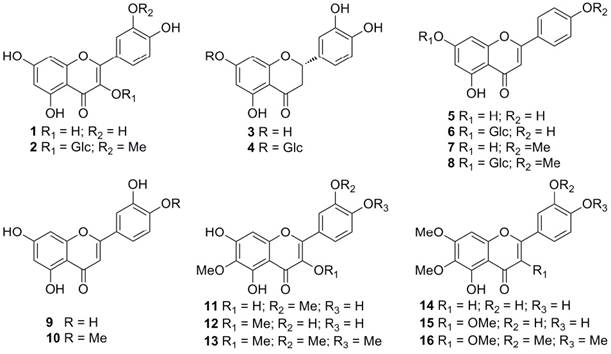
Fig. 1
Flavonoids isolated from the flowers of Chuju
DPPH radical scavenging assay
DPPH assay is a simple and rapid method widely used to measure antioxidant capacity in vitro 26. The antioxidant activity of the flavonoids 1-16 obtained from Chuju was determined by the DPPH radical-scavenging assay and the results were shown in Table 3. Most of the isolated compounds showed fairly strong antioxidant capacity. Quercetin (1) and other six 3’, 4’-dihydroxyl flavonoids (3, 4, 9, 12, 14 and 15) displayed stronger antioxidant activities than that of the positive control ascorbic acid (SC50 = 28.40 μM). Five compounds (2, 5, 6, 10 and 11) exhibited moderate activity with the SC50 values ranged from 51.62 to 130.42 μM. Compounds 7, 8, 13 and 16 appeared to have no activity because their SC50 values exceeded 500 μM.
Antioxidant activity of flavonoids (1-16) from Chuju on DPPH radical-scavenging assay a
Structure-activity relationships (SARs) analysis indicated that the B-ring hydroxylation pattern of flavonoids is the most significant factor for DPPH antioxidant activity. The flavonoids (1, 3, 4, 9, 12, 14 and 15) with an ortho 3’, 4’-dihydroxy mioety in the B-ring possessed much higher antioxidant activity than compounds (2, 5, 6, 10 and 11) with only one free hydroxyl group at C-3’ or C-4’ position in B-ring, due to the B-ring with 3’, 4’-dihydroxy could ensure the capacity for good radical stabilization by delocalization of the unpaired electron in the aroxyl radical structure 27, this trend was further confirmed by the flavonoids (7, 8, 13 and 16) whose hydroxyl groups in B-ring have been substituted by methoxyl groups. Moreover, comparison of the SC50 values of the glucosides (4 and 6) with their aglycones (3 and 5), indicated that the bigger group of O-glucosyl at C-7 had a negative effect on antioxidant activity probaly for the steric hindrance; the same trend of effects were observed for compounds 1 and 2, the O-glucosyl at C-3 in compound 2 was partly responsible for the decrease of its antioxidant activity. The observation is consistent with those reported in previous literature 28,29.
Anti-inflammatory activity
NO is one of the cellular inflammatory mediators of physiological and pathological process 30,31. Stimulation of murine macrophage RAW 264.7 cells by LPS induced inducible nitric oxide synthase (iNOS) and overproduction of NO, which could be quantitative detected by colorimetric method. The results presented in Table 4 revealed that most of the flavonoids isolated from Chuju possessed considerable NO production inhibitory activity. Comparing with the positive control ibuprofen (IC50 = 30.57 μM), nine compounds (1, 5, 7, 9 - 11, 13, 14 and 16) showed stronger activities, 15 displayed moderate activity, and 3, 4 and 12 were inactive. Furthermore, three flavonoid glucosides (2, 6 and 8) showed high cytotoxicity against RAW 264.7 cells treated with LPS (1 μg/ml) in the MTT assay.
Inhibition of flavonoids (1-16) from Chuju on NO production in LPS-activated RAW 264.7 cells
Our investigation demonstrated that Chuju flavonoids effectively inhibited the NO production in LPS-induced RAW 264.7 cells. The positive control, ibuprofen, is a common prescribed drug used to treat various diseases or injures related to inflammation 32,33. In present study, more than half of the flavonoids obtained from Chuju displayed higher inhibition of NO production than the positive control (Ibuprofen, IC50 = 30.57 μM). Comparison of IC50 values between luteolin (9, 15.59 μM) with quercetin (1, 30.01 μM) revealed that flavones had stronger activity than the corresponding flavonols, and this tendency could also be observed from other flavones (5, 7, 9 and 14), which indicated that 3-hydroxyl moiety had a negative impact on the inhibitory effect on NO production 34. The great gap between luteolin (9, 15.59 μM) and eriodictyol (3, > 80 μM) strongly suggested that the anti-inflammatory activity of flavones was higher than those of the corresponding flavanones and the 2-3 double bond in A-ring was structurally necessary 35. Flavonoids 11-13, 15 and 16 were the methoxylated derivatives of quercetagetin with significant amount in Chuju, and the order of their NO inhibiton was 13 > 11> 12 and 16 > 15. These observations showed that the 3’, 4’-dimethoxylations in B-ring tended to enhance the activities. All flavonoid glucosides (2, 4, 6 and 8) were found to be inactive or highly cytotoxic regardless the different types of aglycones, which was in agreement with the report that glycosylation greatly reduced the anti-inflammatory activity 36.
Conclusion
This study demonstrated that Chuju was rich in flavonoids. Bio-guided chemical investigation led the isolation and identification of 16 flavonoids (1-16) from the flowers; compounds 11-16 are first reported for Chrsanthemum species. The flavonoids play an important role in the antioxidant and anti-inflammatory activity for Chuju. The results of this study partially corroborate the traditional uses of Chuju for the treatment of disorders involving oxidative stress and inflammation. From the health point of view, Chuju is an ideal source of natural flavonoids with significant antioxidant and anti-inflammatory effect, and suitable for further development as health-promoting food or supplements.
Experimental
General procedures
Optical rotations were measured on a Jasco P-1020 polarimeter. A Shimadzu UV-2401A spectrophotometer was used to obtain the UV spectra. A Bruker Tensor 27 FT-IR spectrometer was used for IR spectra with KBr pellets. 1D and 2D NMR spectra were recorded on Bruker AM-400 and Avance III-500 spectrometer with TMS as internal standard. Chemical shifts (δ) were expressed in ppm with reference to the solvent signals. MS and HRMS were performed on an API-QSTAR-Pulsar-1 spectrometer.
Chemicals and reagents
DPPH (1,1-Diphenyl-2-picrylhydrazyl), MTT (3-(4,5-cimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide) and L-ascorbic acid was purchased from Sigma-Aldrich (St. Louis, MO, USA). Column chromatography was carried out on Sephadex LH-20 gel (25-100 μM, Pharmacia Fine Chemical Co. Ltd.), MCI gel CHP-20P (75-150 μM, Mitsubishi Chemical Co.), Chromatorex ODS (30-50 μM, Fuji Silysia Chemical Co. Ltd.), and silica gel (200-300 mesh, Qingdao Haiyang Chemical Co. Ltd., China). Thin layer chromatography (TLC) was carried out on silica gel G and H precoated plates (Qingdao Haiyang Chemical Co. Ltd., Qingdao, China) and spots were detected by spraying with 5% H2SO4 in EtOH followed by heating. Other reagents were purchased from Sino-pharm (Shanghai, China).
Plant material
The dried flowers of C. morifolium cv. Chuju were collected in October 2015 from Chuzhou, Anhui Province, China, and identified by Prof. Kai-Jin Wang at School of Life Sciences, Anhui University, where a voucher specimen (No. 2015-10-01) was deposited.
Determination of total flavonoids content (TFC) of Chuju and fractions (A-D)
The total flavonoids content was determined using the AlCl3 method slightly modified 37, and the TFC values were expressed as rutin equivalents (mg RE/g).
Extraction and isolation
The dried flowers of Chuju (5.0 kg) were powered and extracted with 85% EtOH (40 L × 3) at room temperature. After removal of the organic solvent, the extract was concentrated to give a residue (1.0 kg). The crude extract was suspended in H2O, and then passed through a D101 macroporous resin column eluted with water followed by 30%, 60% and 100% MeOH to yield four fractions (A-D). Fractions C and D showed the higher flavonoid content in TFC test; fraction C exhibited the strongest antioxidant and anti-inflammatory activities, and fraction D displayed moderate activities. Thus, further chemical investigation was focused on the two flavonoids-enriched fractions (C and D).
The fraction C (200 g) was fractionated on silica gel column chromatography (CC) eluted with CHCl3-MeOH (50:1-2:1) to give six fractions (C1-C6). Repeated column chromatography on Sephadex LH-20 and ODS, eluted with H2O-MeOH (1:0-0:1), led the isolation of compound 5 (50 mg) from fraction C1 (5.6 g), and compounds 10 (400 mg) and 11 (30 mg) from fraction C2 (3.9 g). Fraction C3 (6.8 g) was applied on MCI gel CHP 20P column eluted with H2O-MeOH (1:0-0:1) to give to three fractions (C3-1-C3-3), fraction C3-1 (720 mg) was first purified on Sephadex LH-20 eluted with EtOH, followed by ODS CC eluted with H2O-MeOH (1:0.3-1:1) to yield compound 12 (40 mg). Fraction C4 (8.1 g) was divided into five fractions (C4-1-C4-5) by MCI gel CHP 20P column chromatography H2O-MeOH (1:0-0:1), and repeated column chromatography on ODS, eluted with EtOH followed by H2O-MeOH (1:0.3-1:1), led the isolation of compound 3 (80 mg) from fraction C4-1 (1.1 g), and 1 (35 mg) from fraction C4-2 (4.2 g). Fraction C5 (30.6 g) was subjected on silica gel eluted with CHCl3-MeOH (10:1-3:1) to give four fractions (C5-1-C5-4), fraction C5-1 (1.2 g) was purified on Sephadex LH-20, ODS and MCI gel CHP 20P to achieve compound 2 (10 mg), fraction C5-2 (3.7 g) was insoluble in MeOH to get the precipitate of pure 8 (2.0 g). fraction C5-4 (23.7 g) was also insoluble in MeOH to get the precipitate of pure 13 (12.0 g).Fraction C6 (12.6 g) was submitted on MCI gel CHP 20P column eluted with H2O-MeOH (1:0.3-1:1), and purified by Sephadex LH-20 eluted with MeOH to yield compounds 4 (30 mg), 7 (15 mg) and 9 (2.0 g).
The fraction D (150 g) was fractionated on silica gel CC eluted with petroleum ether-acetone (60:1-7:1) to give five fractions (D1-D5). Fraction D1 35.0 g) was subjected on silica gel CC eluted with petroleum ether-EtOAc (10:1-1:1) to afford two fractions (D1-1 and D1-2). Fraction D1-1 (5.2 g)was applied on Sephadex LH-20 CC eluted with EtOH, followed by preparative TLC (CHCl3-acetone, 7:1) to give compounds 15 (208 mg), and 16 (150 mg). Fraction D2 was chromatographed on MCI gel CHP 20P column eluted with H2O-MeOH (1:0-0:1) to afford two fractions (D2-1 and D2-2), further purified fraction D2-1 on ODS (60-80% MeOH), followed by Sephadex LH-20 CC (EtOH) to yield compounds 6 (20 mg) and 14 (16 mg).
Quercetin (1). yellow amorphous powder, negative ESI-MS m/z: 301 [M-H]-; 1H-NMR (400 MHz, DMSO-d 6) δ: 12.50 (1H, s, OH-5), 6.18 (1H, d, J = 2.0 Hz, H-6), 6.40 (1H, d, J = 2.0 Hz, H-8), 7.67 (1H, d, J = 2.0 Hz, H-2’), 6.88 (1H, d, J = 8.0 Hz, H-5’), 7.54 (1H, dd, J = 2.0, 8.0 Hz, H-6’); 13C-NMR (100 MHz, DMSO-d 6): see Table 2.
Isorhamnetin 3-Ο-β-D-glucoside (2). yellow amorphous powder, negative ESI-MS m/z: 477 [M-H]-; 1H-NMR (400 MHz, DMSO-d 6) δ: 12.60 (1H, s, 5-OH), 6.17 (1H, s, H-6), 6.39 (1H, s, H-8), 7.94 (1H, s, H-2’), 7.48 (1H, d, J = 8.0 Hz, H-6’), 6.92 (1H, d, J = 8.0 Hz, H-5’), 3.80 (3H, s, 3-OCH3), 5.57 (1H, d, J = 8.0 Hz, Glc: H-1’’): see Table 2.
Eriodictyol (3). light yellow amorphous powder, negative ESI-MS m/z: 287 [M-H]-; 1H-NMR (400 MHz, DMSO-d 6) δ 12.15 (1H, s, 5-OH), 5.37 (1H, dd, J = 2.8, 12.0 Hz, H-2), 3.17 (1H, dd, J = 12.0, 16.4 Hz, H-3a), 2.66 (1H, dd, J = 2.8, 16.4 Hz, H-3b), 5.88 (2H, s, H-6, 8), 6.87 (1H, s, H-2), 6.74 (2H, s, H-5’, 6’); 13C-NMR (100 MHz, DMSO-d 6): see Table 2.
Pyracanthoside (4). light yellow amorphous powder, negative ESI-MS m/z: 449 [M-H]-; 1H-NMR (400 MHz, DMSO-d 6) δ: 12.09 (1H, s, 5-OH), 5.43 (1H, dd, J = 3.0, 12.6 Hz, H-2), 3.17 (1H, dd, J = 12.6, 16.0 Hz, H-3a), 2.73 (1H, dd, J = 3.0, 16.0 Hz, H-3b), 6.14 (1H, d, J = 2.0 Hz, H-6), 6.15 (1H, d, J = 2.0 Hz, H-8), 6.89 (1H, br s, H-2’), 6.76 (2H, br s, H-5’, 6’), glc: 5.11 (1H d, J = 8.0 Hz, H-1’’; 13C-NMR (100 MHz, DMSO-d 6): see Table 2.
Apigenin (5). yellow amorphous powder, negative ESI-MS m/z: 269 [M-H]-; 1H-NMR (400 MHz, DMSO-d 6) δ: 12.90 (1H, s, 5-OH), 6.72 (1H, s, H-3), 6.12 (1H, d, J = 2.0 Hz, H-6), 6.41 (1H, d, J = 2.0 Hz, H-8), 7.96 (2H, d, J = 8.0 Hz, H-2’, 6’), 6.95 (2H, d, J = 8.0 Hz, H-3’, 5’). 13C-NMR (100 MHz, DMSO-d 6): see Table 2.
Apigetrin (6). yellow amorphous powder, negative ESI-MS m/z: 431 [M-H]-; 1H-NMR (400 MHz, DMSO-d 6) δ: 12.99 (1H, s, 5-OH), 6.88 (1H, s, H-3), 6.44 (1H, d, J = 2.0 Hz, H-6), 6.82 (1H, d, J = 2.0 Hz, H-8), 7.96 (2H, d, J = 8.0 Hz, H-2’, 6’), 6.94 (2H, d, J = 8.0 Hz, H-3’, 5’), glc: 5.07 (1H, d, J = 7.2 Hz, H-1’’); 13C-NMR (100 MHz, DMSO-d 6): see Table 2.
Acacetin (7). light yellow amorphous powder, negative ESI-MS m/z: 283 [M-H]-; 1H-NMR (500 MHz, C5D5N) δ: 7.92 (2H, d, J = 8.8 Hz, H-2’, 6’), 7.04 (2H, d, J = 8.8 Hz, H-3’, 5’), 6.92 (1H, s, H-3), 6.87 (1H, d, J = 1.6 Hz, H-8), 6.53 (1H, d, J = 1.6 Hz, H-6), 3.81 (3H, s, 4’-OCH3); 13C-NMR (125 MHz, C5D5N): see Table 2.
Acacipetalin (8). light yellow amorphous powder, negative ESI-MS m/z: 445 [M-H]-; 1H-NMR (400 MHz, DMSO-d 6) δ: 12.93 (1H, s, 5-OH), 8.07 (2H, d, J = 8.0 Hz, H-2’, 6’), 7.13 (2H, d, J = 8.0 Hz, H-3’, 5’), 6.98 (1H, s, H-3), 6.46 (1H d, J = 2.0 Hz, H-6), 6.86 (1H, d, J = 2.0 Hz, H-8), 3.86 (3H, s, 4’-OCH3), glc: 5.08 (1H, d, J = 8.0 Hz, H-1’’); 13C-NMR (100 MHz, DMSO-d 6): see Table 2.
Luteolin (9). yellow amorphous powder, negative ESI-MS m/z: 285 [M-H]-; 1H-NMR (400 MHz, acetone-d 6) δ: 12.97 (1H, s, 5-OH),6.67 (1H, s, H-3), 6.21 (1H, d, J = 2.0 Hz, H-6), 6.46 (1H, d, J = 2.0 Hz, H-8), 7.40 (1H, d, J = 2.0 Hz, H-2’), 6.90 (1H, d, J = 8.0 Hz, H-5’), 7.46 (2H, dd, J = 2.0, 8.0 Hz, H-6’). 13C-NMR (100 MHz, acetone-d 6): see Table 2.
Diosmetin (10). yellow amorphous powder, negative ESI-MS m/z: 299 [M-H]-; 1H-NMR (400 MHz,DMSO-d 6) δ: 12.93 (1H, s, 5-OH), 6.64 (1H, s, H-3), 6.19 (1H, d, J = 1.8 Hz, H-6), 6.50 (1H, d, J = 1.86 Hz, H-8), 7.42 (1H, d, J = 2.2 Hz, H-2’), 7.03 (1H, d, J = 8.5 Hz, H-5’), 7.53 (1H, dd, J = 2.2, 8.5 Hz, H-6’), 3.86 (3H, s, 4’-OCH3); 13C-NMR (100 MHz, DMSO-d 6): see Table 2.
Spinacetin (11). yellow amorphous powder, negative ESI-MS m/z: 345 [M-H]-; 1H-NMR (400 MHz, acetone-d 6) δ: 13.00 (1H, s, 5-OH), 6.56 (1H, s, H-8), 7.71 (1H, d, J = 2.0 Hz, H-2’), 6.99 (1H, d, J = 8.4Hz, H-5’), 7.58 (1H, dd, J = 2.0, 8.4 Hz, H-6’), 3.87 (3H, s, -OCH3), 3.86 (3H, s, -OCH3); 13C-NMR (100 MHz, acetone-d 6): see Table 2.
Axillarin (12). yellow amorphous powder, negative ESI-MS m/z: 345 [M-H]-; 1H-NMR (400 MHz, DMSO-d 6) δ: 12.78 (1H, s, 5-OH), 6.46 (1H, s, H-8), 7.52 (1H, d, J = 2.0 Hz, H-2’), 6.89 (1H, d, J = 8.4 Hz, H-5’), 7.43 (1H, dd, J = 2.0, 8.4 Hz, H-6’), 3.77 (3H, s, -OCH3), 3.73 (3H, s, -OCH3); 13C-NMR (100 MHz, DMSO-d 6): see Table 2.
Bonanzin (13). yellow amorphous powder, negative ESI-MS m/z: 373 [M-H]-; 1H-NMR (400 MHz, acetone-d 6) δ: 7.70 (1H, d, J = 2.0 Hz, H-2’), 7.68 (1H, dd, J = 2.0, 8.4 Hz, H-6’), 7.09 (1H, d, J = 8.4 Hz, H-5’), 6.45 (1H, s, H-8), 3.89 (3H, s, -OCH3), 3.88 (3H, s, -OCH3), 3.85 (3H, s, -OCH3), 3.84 (3H, s, -OCH3); 13C-NMR (100 MHz, acetone-d 6): see Table 2.
Cirsiliol (14). light yellow amorphous powder, negative ESI-MS m/z: 329 [M-H]-; 1H-NMR (500 MHz, C5D5N) δ: 13.74 (1H, s, 5-OH), 6.99 (1H, s, H-3), 6.70 (1H, s, H-8), 7.60 (1H, d, J = 1.8 Hz, H-2’), 7.38 (1H, d, J = 7.9 Hz, H-5’), 7.99 (1H, dd, J = 1.8, 7.9 Hz, H-6’), 4.01 (3H, s, OCH3), 3.89 (3H, s, OCH3); 13C-NMR (125 MHz, C5D5N): see Table 2.
Chrysosplenol D (15). light yellow amorphous powder, negative ESI-MS m/z: 359 [M-H]-; 1H-NMR (500 MHz, CD3OD) δ: 12.53 (1H, s, 5-OH), 6.67 (1H, s, H-8), 7.61 (1H, d, J = 1.8 Hz, H-2’), 6.87 (1H, d, J = 7.3 Hz, H-5’), 7.50 (1H, dd, J = 1.8, 7.3 Hz, H-6’), 3.91 (3H, -OCH3), 3.79 (3H, -OCH3), 3.75 (3H, -OCH3); 13C-NMR (125 MHz, CD3OD): see Table 2.
Artemetin (16): light yellow amorphous powder, negative ESI-MS m/z: 387 [M-H]-; 1H-NMR (500 MHz, C5D5N) δ: 13.35 (1H, s, 5-OH), 6.89 (1H, s, 8-H), 7.90 (1H, d, J = 2.0 Hz, H-2’), 7.14 (1H, d, J =8.5 Hz, H-5’), 7.94 (1H, dd, J = 2.0, 8.5 Hz, H-6’), 4.05 (3H, s, OCH3), 4.04 (3H, s, -OCH3), 3.89 (3H, s, -OCH3), 3.89 (3H, s, -OCH3), 3.88 (3H, s, -OCH3); 13C-NMR (125 MHz, C5D5N): see Table 2.
13C-NMR (δ in ppm) data of compounds 1-16
DPPH radical-scavenging assay
The DPPH radical-scavenging assay was performed as previously described 38. In this assay, L-ascorbic acid was used as the positive control, and reaction mixtures containing an ethanolic solution of 200 μM DPPH (100 μL) and two-fold serial dilutions of sample (dissolved in 100 μL ethanol, with amounts of sample ranging from 2 to 1000 μg/mL) were placed in a 96 well microplate and incubated at 37°C for 30 min. After incubation the absorbance was read at 517 nm by an Emax precision microplate reader and the mean of three readings was obtained. Scavenging activity was determined by the following equation: % scavenging activity = [Acontrol-Asample]/Acontrol × 100. The SC50 values were obtained through extrapolation from linear regression analysis and denoted the concentration of sample required to scavenge 50% of DPPH radicals.
Measurement of nitric oxide production
The anti-inflammatory ability on inhibition of NO production in LPS-induced RAW 264.7 cells was performed according to Ma 39. In brief, murine macrophage cell line RAW 264.7 was purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). RAW 264.7 cells were cultured in DMEM medium (Thermo Fisher Scientific, USA) supplemented with 10% FBS (Sijiqing, China) at 37°C in a humidified atmosphere with 5% CO2. Ibuprofen was used as the positive control. The nitrite concentration was measured in supernatants of cultured RAW 264.7 cells according to the Griess reaction as an indicator of NO production by using an NO assay kit (Beyotime Insititute of Biotechnolgy, China). Briefly, RAW 264.7 cells at 5×103 per well were seeded in 96-well plates. Cells were seeded in 96-well culture plates for 12h incubation, and then pretreated with tested compounds (3 to 50 µM) and LPS (Sigma, USA, 1 µg/mL in PBS) for 24h. 50 μL of cell-free supernatant was mixed with 100 μL of Griess reagent (1% sulfanilamide, 0.1% naphthylethylenediamine dihydrochloride, and 2.5% phosphoric acid) and incubated at room temperature for 5min. The concentration of nitrite was measured at 540 nm. Sodium nitrite (NaNO2) was used as a standard to calculate the NO concentration.
MTT assay
The cell viability was determined by MTT method as previously described by Ma 39. The cell viability was determined by MTT method. In brief, RAW 264.7 cells at 5×103 per well were seeded in 96-well plates. After 12 h incubation, the cells were treated with or without tested compounds for 24 h. Then, 20 µL MTT solutions (5 mg/ml) were added to each well for another 4 h and the resulting formazan crystals were dissolved in DMSO. The optical density was measured at 490 nm. The cytotoxicity was calculated from the plotted results using untreated cells at 100%.
Statistics
All the experiments were performed in three replications, and data were expressed as means ± SD of three independent experiments.
Acknowledgements
This research was supported financially by Zhenxing Plan of Education Department of Anhui Province (KJ2016SD03) and Collaborative Innovation Center of Modern Bio-manufacture, Anhui University (Y01008410). The authors are grateful to the Modern Experiment Technology Center, Anhui University for measuring of 400 and 500 MHz NMR, and other spectral data.
References
1. 1. Pisoschi, A. M.; Pop, A. Eur. J. Med. Chem. 2015, 97, 55-74.
2. 2. Lei Y.; Wang K.; Deng L, Chen Y, Nice E. C.; Huang C. Med. Res. Rev. 2015, 35, 306-340.
3. 3. Young I. S.; Woodside J.V. J. Clin. Pathol. 2001, 54, 176-186.
4. 4. Sharla, R. in: Antioxidants, The truth about BHA, BHT, TBHQ and other antioxidants used as food additives. 1sted. Tigmor Books, Northumberland, UK, 2009, 19-30.
5. 5. Williams G. M.; Iatropoulos M. J.; Whysner J. Food Chem. Toxicol. 1999, 37, 1027-1038.
6. 6. Pietta P. G. J. Nat. Prod. 2000, 63, 1035-1042.
7. 7. Editorial Board of Zhong-Hua-Ben-Cao, in: Zhong-Hua-Ben-Cao, Vol. 21, Shanghai Science and Technology Press, Shanghai, China, 1999, 806-810.
8. 8. He D. X.; Ru X. C.; Wen L.; Wen Y. C.; Jiang H. D.; Bruce I. C.; Jin J.; Ma X.; Xia Q. J. Ethnopharmacol. 2012, 139, 68-73.
9. 9. Ukiya M.; Akihisa T.; Yasukawa K.; Kimura Y.; Koike K.; Nikaido T.; Nikaido T. ; Takdo M. J. Agr. Food Chem. 2001, 49, 3187-3197.
10. 10. Sugawara T.; Igarashi K. Food Sci. Technol. Res. 2009, 15, 499-506.
11. 11. Ukiya M. ; Akihisa T. ; Tokuda H.; Suzuki H.; Mukainaka T.; Ichiishi E.; Yasukawa K. ; Kasahara Y.; Nishino H. Cancer Lett. 2002, 177, 7-12.
12. 12. Wang M.; Avula B.; Wang Y. H.; Zhao J. P.; Cristina Avonto C.; Parcher J. F.; Raman V.; Zweigenbaum J. A.; Wylie P. L.; Khan I. A. Food Chem. 2014, 152, 391-398.
13. 13. Wang Y. J.; Guo Q. S.; Yang X. W.; Xu W. B.; Tao H. Y. Chin. J. Chin. Mater. Med. 2008, 33, 2207-2211.
14. 14. Lin L. Z.; Harnly J. M. (2010). Food Chem. 2010, 120, 319-326.
15. 15. Clifford M. N.; Wu W. G.; Kirkpatrick J.; Kuhnert N. J. Agr. Food Chem., 2007, 55, 929-939.
16. 16. Qin J. J.; Zhu J. X.; Zhu Y.; Jin H. Z.; Lv Y. H.; Zhang W. D. Chin. J. Nat. Med., 2010, 8, 257-259.
17. 17. Kong D. Y.; Luo S. Q.; Li T. H.; Lei X. H. Acta Pharm. Sin. 1988, 23, 593-600.
18. 18. Pan Y. G.; Li Y. X.; Yuan M. Q. Food Chem. 2015, 186, 119-122.
19. 19. Zhang Y. J.; Abe T.; Tanaka T.; Yang C. R.; Kouno I. (2002). Chem. Pharm. Bull. 2002, 50, 841-843.
20. 20. Bensouici C.; Kabouche A.; Karioti A.; Oztürk M.; Duru M. E.; Billa A. R.; Kabouche Z. Pharm. Bio. 2015, 7, 1-6.
21. 21. Zou Z. J.; Ju J. H.; Yang J. S. Chin. Pharm. J. 2006, 41, 102-103.
22. 22. Park Y.; Moon B.H.; Yang H.; Lee H.; Lee E.; Lim Y. Magn. Reson. Chem. 2007, 45, 1072-1075.
23. 23. Tang, H. Q.; Hu, J.; Yang, L.; Tan, R. X. Planta Med. 2000, 66, 391-393.
24. 24. Morita, N.; Shimizu, M.; Arisawa, M. Phytochemistry 1973, 12, 421-423.
25. 25. Zhao, Y. W.; Ni, F. Y.; Song, Y. L.; Wang, S. Y.; Huang, W. Z.; Wang, Z. Z.; Xiao, W. Chin. J. Chin. Mater. Med. 2014, 39, 4816-4821.
26. 26. Blois, M. S. Nature 1958, 181, 1199-1200.
27. 27. Van Acker, S. A.; de Groot, M. J.; van, D. J.; Tromp, M. N.; den Kelder, G. D.; van der Vijgh, W. J. F.; Bast, A. Chem. Res. Toxicol. 1996, 9, 1305-1312.
28. 28. Braca, A.; Fico, G.; Morelli, I.; Simone, F. D.; Tome, F.; Tommasi, N. D. J. Ethnopharmacol. 2003, 86, 63-67.
29. 29. Cioffi, G.; D’Auria, M.; Braca, A.; Mendez, J.; Castillo, A.; Morelli, I.; De Simone, F.; De Tommasi, N. J. Nat. Prod. 2002, 65, 1526-1529.
30. 30. Moncada, S.; Palmer, R. M. J.; Higgs, E. A. Pharmacol. Rev. 1991, 43, 109-142.
31. 31. Nathan C. FASEB J. 1992, 6, 3051-3064.
32. 32. Andrews, J.; Djakiew, D.; Krygier, S.; Andrews, P. Cancer Chemother. Pharmacol. 2002, 50, 277-284.
33. 33. Tsai, W. C.; Tang, F. T.; Hsu, C. C.; Hsu, Y. H.; Pang, J. H.; Shiue, C. C. J. Orthop. Res. 2004, 22, 586-591.
34. 34. Matsuda, H.; Morikawa, T.; Ando, S. I.; Yoshikawa, M. Bioorg. Med. Chem. 2003, 11, 1995-2000.
35. 35. Panthong, A.; Kanjanapothi, D.; Tuntiwachwuttkul, P.; Pancharoen, O.; Reutrakul, V. Phytomedicine 1994, 1, 141-144.
36. 36. Kim, H. K.; Cheon, B. S.; Kim, Y. H.; Kim, S. Y.; Kim, H. P. Biochem. Pharmacol. 1999, 58, 759-765.
37. 37. Liao, W. C.; Lai, Y. C.; Yuan, M. C.; Hsu, Y. L.; Chan, C. F. Food Chem. 2001, 127, 1224-1228.
38. 38. Wang, K. J.; Yang, R. Y.; Zhang Y. J. Food Chem. 2007, 101, 365-371.
39. 39. Ma, W.; Liu, F.; Ding, Y. Y.; Zhang, Y.; Li, N. Fitoterapia 2015, 105, 83-88.
Author notes
*Corresponding authors E-mail address: 1965475452@qq.com (N. Li), wkjahla@163.com (K. J. Wang)
