Journal of the Mexican Chemical Society, vol. 61, no. 2, 2017
Sociedad Química de México A.C.
María Modroño-Alonso
Universidad de Carabobo, Venezuela
William Castro
Instituto Venezolano de Investigaciones Científicas, Venezuela
Francisco Lopez-Linares
Chevron Energy Technology Company, United States of America
Merlín Rosales
Universidad del Zulia, Venezuela
Pablo Jose Baricelli *
Universidad de Carabobo, Venezuela
Date received: 18 October 2016
Date accepted: 08 March 2017
Funding
Funding source: FONACIT-VENEZUELA
Contract number: F-97003766
Contract number: CDCH-UC project 94017
Abstract.: The water soluble cobalt complex HCo(CO)[P(o-C6H4SO3Na)]3 was used as catalyst precursor for the biphasic aqueous hydroformylation of 1-hexene. The complex was synthesized by reductive carbonylation of CoCl2.6H2O in the presence of o-TPPTS ([P(o-C6H4SO3Na)]3) under nitrogen atmosphere and characterized by FTIR, 1H NMR and 31P {1H} NMR, 13C NMR, DEPT - 135, COSY, HSQC, MS (ESI). The catalytic activity of the complex in the biphasic hydroformylation reaction of 1-hexene was evaluated under moderate reaction conditions. The pressure and temperature were varied from 4137 - 7584 kPa (600-1100 psi) of syngas and from 353 - 383 K (80 - 110 oC), respectively. The 1-hexene concentration was varied from 0.021-0,11M and the catalyst from 4x10-4 - 1.1x10-3 M. The best conversion at 363 K and 7584 kPa and 7.5 h was 62% with selectivity towards aldehydes (heptanal and 2-methyl-hexanal) of 66% to with l/b ratio of 2.6. The recycling of the catalytic precursor after four successive times, did not show any loss on the activity, having selectivity towards aldehyde up to 60%.
Keywords: Biphasic hydroformylation, Cobalt complexes, 1-hexene, recycling, o-TPPTS.
Resumen.: El complejo soluble en agua HCo(CO)[P(o-C6H4SO3Na)]3 fue usado como precursor catalítico para la hidroformilación de 1-hexeno. El complejo fue sintetizado por carbonilación reductiva de CoCl2.6H2O en presencia de o-TFFTS,([P(o-C6H4SO3Na)]3) en atmosfera de nitrógeno y caracterizado por IRFT 1H RMN y 31P {1H} RMN, 13C RMN, DEPT - 135, COSY, HSQC, MS (ESI). La actividad catalítica del complejo en la hidroformilación bifásica acuosa de1-hexeno fue evaluada bajo condiciones moderadas de reacción. La presión y la temperatura se variaron desde 4137 - 7584 kPa (600-1100 psi) de gas de síntesis y de 353 - 383 K (80 - 110 oC), respectivamente. La concentración de 1-hexeno se varió desde 0.021-0.11M y la del catalizador desde 4x10-4 - 1.1x10-3 M. La mejor conversión fue 62% a 363 K y 7584 kPa en 7.5 h con una selectividad de 66% hacia los aldehídos (heptanal y 2-metil-hexanal), obteniéndose y una relación n/i de 2.6. El precursor catalítico fue reciclado durante cuatro veces sucesivas sin mostrar perdida en la actividad catalítica y con una selectividad hacia aldehídos de 60 %.
Palabras clave: Hidroformilación bifásica, complejos de cobalto, 1-hexeno, reciclo, o-TFFTS.
Introduction
Since the initials work by Kuntz [1] and the development work at Ruhrchemie/Rhone-Poulenc process for the hydroformylation of propylene to butyraldehyde [2-5] the oxo process continue to be the more important industrial application of homogeneously catalyzed reactions by transition complexes [6-9]. However, the cost of the rhodium complexes and the use of surfactants and other techniques [10-13] to improve the solubility of the molecular weight olefins indeed made the biphasic process in some cases to expensive. For this reasons, we are returned with the use of cobalt complexes in two phase hydroformylation of olefins. The hydroformylation reactions catalyzed by cobalt complexes are not a new matter. In the 50’s decade was reported the first generation of non-modified cobalt catalyst, development and implemented by BASF and EXXON. In the 60’s decade, the phosphines were used as stabilizing ligands and the active specie was the HCo(CO)3(PR3). However, during the 70’s and 80’s the rhodium replaced the cobalt complexes and the active species were the HRh(CO)(PPh3)3 used by the Union Carbide Corporation and HRh(CO)(TPPTS)3 used by the Ruhrchemie/Rhôme-Poulenc [14]. Nevertheless, in the industrial processes, uses of cobalt complexes in their second generation are the appropriate complexes for the economic production of long chain aldehydes and alcohols. In 2012, the annual productions of oxo products was eighth million tons and where around one million continue to be produce using cobalt complexes [14].
Because of the elevated cost of the precious metals and it incidence in the final catalyst cost, in the last years, it has been an increased number of publications using cobalt catalyst with TPPTS as catalytic precursors in the biphasic hydroformylation reactions. Beller and co-workers [15] reported the hydroformylation of pentenes at high temperature with Cobalt/TPPTS system which reach an l/b ratio of 70/30 and a without loss of activity after four time recycling. Parmar and co-workers [16], reported the synthesis of the CoCl2(TPPTS)2 and the evaluation of it activity in the hydroformylation of 1-hexene at 100oC and 1300 psi of syngas. Dabbawala and co-workers [17] studied the catalytic activity of the CoCl2(TPPTS)2 complex in the hydroformylation of 1-octene and 1-decene at 100 oC and 1160 psi off syngas. The use of surfactant such as cetyltrimethyl ammonium chloride (CTAB) increases the activity of the complex as well as the aldehyde selectivity. Also by using an excess of TPPTS; 8:1 TPPTS/Co, the migration of the cobalt to the organic phase was suppressed completely.
Recently, our group reported the in situ catalytic activity of the system Co/TPPMS in the hydroformylation reaction of 1-hexene at 100 oC; 1000 psi; 0.0005 mmol of Co; 0.5 mmol of 1-hexene; 8 mmol of TPPMS; 1/1 CO/H2; 1020 rpm, where 95% of total conversion with 83% of aldehyde selectivity was achieved after 8h of reaction [18]. It is interesting that most of the recent cobalt reports has been focused in CoCl2(TPPTS)2 and the hydride species like CoH(CO)(TPPTS)3 was reported by Herrmann and co-workers in the 90’s [19]. For this reason, we decided to investigate the potentiality of the complex [CoH(CO)(TPPTS)3, considering that the sulfonate group is located in ortho position, as catalyst precursor for the aqueous biphasic hydroformylation reaction of 1-hexene. Evaluations of the operational conditions in details are presented.
Results and discussion
Synthesis and characterization of trisodium salt of 2,2´,2´´-phosphinetriylbenzene-sulfonic acid (o-TPPTS)
The TPPTS tri(o-sulfonatophenyl)phosphine was prepared according to the modified methods reported in the literature [20]. The original version was designed for the synthesis of tri(m-sulfonatophenyl)phosphine. However, when the temperature of the sulfonation reaction was kept between 278 - 283K (5-10°C) instead of 293 - 295 K (20-22°C), the pure tri(o-sulfonatophenyl)phosphine was obtained. On the other hand, all NMR signals were unequivocally assigned on the basis of 1D and 2D, correlation spectroscopy (COSY) and heteronuclear single quantum correlation (HSQC) experiments (for complete NMR data see experimental section). The spectrum of o-TPPTS in D2O shows two triplets centered at 7.35 ppm and 7.44 ppm and two doublets centered at 7.70 ppm and 7.77 ppm. This pattern is consistent with a sulfonation of the phosphine in orto position as consequence of the reaction conditions employed such as time: 150 hours, temperature of 278 - 283 K (5 - 10°C) and without stirring.
The 31P {1H} (Fig. 2) shows only one singlet at -5.76 ppm corresponding to the tri(o-sulfonatophenyl)phosphine. Additionally, it shows the highly purity of the ligand (100% pure) and high efficiency of the washing method in reproducible way.
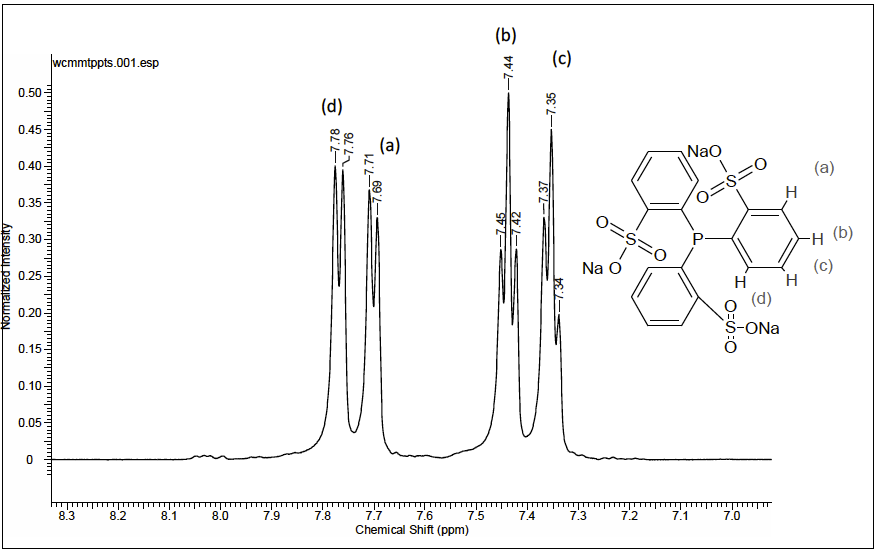
Fig. 1
1H NMR spectrum for the ligand o-TPPTS in D2O at 298K (25°C).
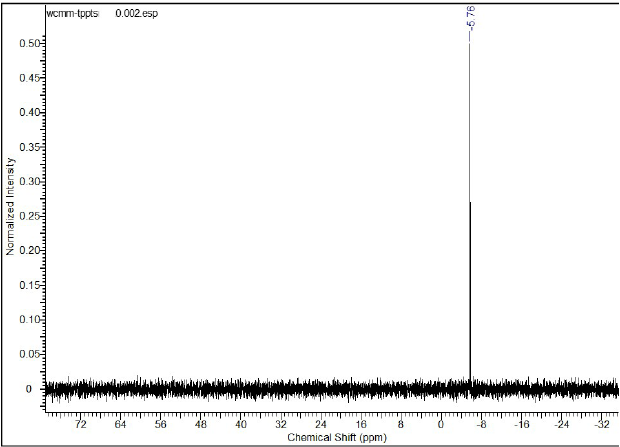
Fig. 2
31P {1H} NMR spectrum for the ligand o-TPPTS in D2O at 298K (25°C).
According to the available data (FT-IR, NMR and MS (ESI)), all data support the sulfonation in ortho position for this ligand.
Synthesis and characterization of HCo(CO) [P(o-C6H4SO3Na)]3 complex.
The cobalt complex was prepared according to a modified method reported previously by Hermann et al. [19]. The FT-IR spectrum (KBr disk) of the complex (Fig. 3) shows sharp bands at 1956 cm-1 and 1905 cm-1 characteristic of hydride and carbonyl groups respectively. Also three sulfonating bands at 1098 cm-1; 1146 cm-1 and 1198 cm-1 assigned to SO3 group from the o-TPPTS and one strong band at 1637 cm-1 assigned to aromatic C=C bond. Similar results were reported by Hermann et al. [19]: two bands at 1953 cm-1 and 1904 cm-1 identified as hydride and carbonyl bands respectively.
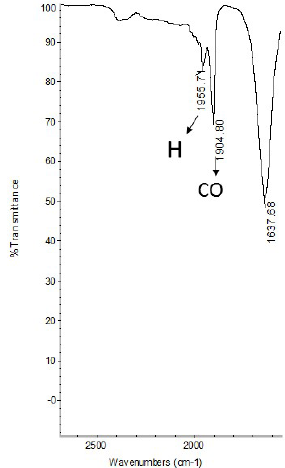
Fig. 3
FTIR for the complex HCo(CO)[P(o-C6H4SO3Na)]3 in KBr.
All NMR signals were unequivocally assigned on the basis of 1D and 2D, correlation spectroscopy (COSY) and heteronuclear single quantum correlation (HSQC) experiments (for complete NMR data see experimental section). Specifically in the 1H NMR (D2O) for the complex, shows a series of signals (Fig. 4) corresponding to the hydrogen from o-TPPTS ligand between 7.06 and 7.90 ppm (m, 36H). Also a single hydride signal, was observed at -12.40 ppm (q, 1H), which is the product of the coupling between the hydride and the three equivalents phosphorous atoms of the o-triphenylphosphine trisulfonate, similar to the one reported by Herrmann et al.[19] centered at -12.46 ppm.
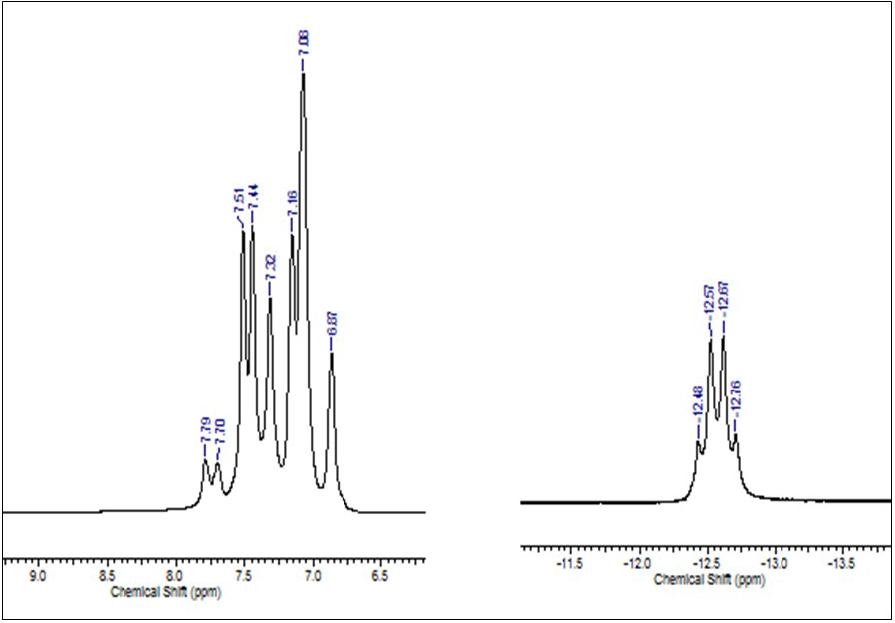
Fig. 4
1H NMR spectrum for the complex HCo(CO)[P(o-C6H4SO3Na)]3 in D2O at 298K (25°C).
The 31P {1H} Fig. 5 shows one singlet at 57.75 ppm assigned to three equivalents phosphines bond to the cobalt, similar to those reported by Bartik at el [21] for cobalt carbonyls complexes with TPPTS, one signal of free o-triphenylphosphine trisulfonate at -5.05 ppm (s, 1P) and one non- signal at 20,93 ppm that is associated to the o-triphenylphosphine oxide formed during the synthesis [20a]. Based from the 31P NMR, the purity of the complex was calculated to be around 81.56% whereas free phosphine and the corresponding oxide were around 9.15 % and 9.29 % respectively. With all information (FT-IR, NMR and MS (ESI)), it suggests a trigonal bipyramidal structure of the complex with three phosphines in the equatorial plane and the hydride and carbonyl in the axial position.
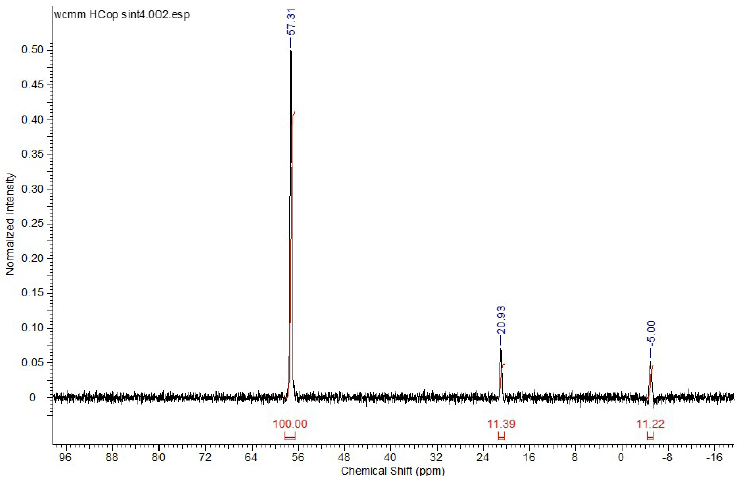
Fig. 5
31P {1H} NMR spectrum for the complex HCo(CO)[P(o-C6H4SO3Na)]3 in D2O at 298K (25°C).
Biphasic catalytic reactions
In order to stablish the best operational conditions for the biphasic hydroformylation reaction of 1-hexene (scheme 1). Experiments were conducted at different pressure and temperature, substrate and catalyst concentrations having a fix reaction time of 7.5 h.
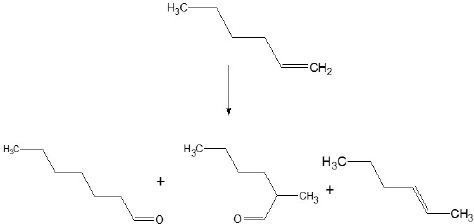
Scheme 1
Products characterized for the hydroformylation of 1 - hexene.
Effect of temperature and syngas pressure
Table 1 shows the influence of the syngas pressure at 373 K (100°C) and 383 K (110oC). The first observation is the absence of hydrogenation products and the only formation of 2-hexenes as isomerization product and secondly the decrease in the induction period of the reaction with the increment of syngas pressure.
Effect of pressure and temperature on the hydroformylation of 1-hexene, catalyzed by HCo(CO)[P(o-C6H4SO3Na)]3 in aqueous medium.

Also, as it is observed in Table 1, 383 K (110oC) does not improve the activity or selectivity of the complex to aldehydes, except at high pressures (7584 kPa =1100 psi), while at 373K (100oC) from 4137 kPa to 6895 KPa (600 to 1000psi) decrease the activity to aldehydes, at 7584 kPa the activity tends to increase, presumably, due to a change of active specie in the reaction media. For these reasons, we can deduce that the increase in the syngas pressure produce the better solubility of the gases in water and also the formation of more active catalytic species than others ran at lower pressure.
In Fig. 6, it can be observed that the activity of the catalyst at temperature between 353 - 383 K (80 - 110 oC), follow a volcano behavior at 5516 kPa (800psi) (6b) and 7584 kPa (1100 psi) (6a) of syngas with the maxima activity at 363 K (90 oC) in both cases.
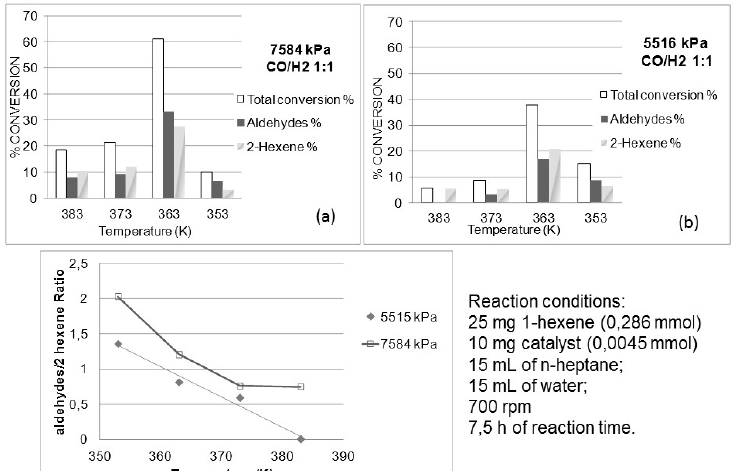
Fig. 6
Temperature influence at 7584 kPa (a); 5516 kPa (b); aldehydes/2-hexene ratio (c).
At 353 K (80oC), the aldehyde selectivity is highly favored but the conversion towards aldehydes was reduced more than 20 %. Also, the activity of the catalyst and the selectivity to aldehyde increase at 363 K (90oC) with the increase of syngas pressure; establishing 363K and 7584 kPa of syngas as the more favorable conditions for the biphasic aqueous hydroformylation reaction of 1-hexene.
Effect of the substrate / catalyst ratio
In the same order of ideas, the increases of the substrate/catalyst ratio, decreases on 1-hexene conversion maintaining the products distribution, is presented in Fig. 7a at 363 K (90oC) and 7584 kPa (1100 psi). These results are expected, because we have the same amount of catalyst for larger moles of substrate required to be transformed at the same interval of time.
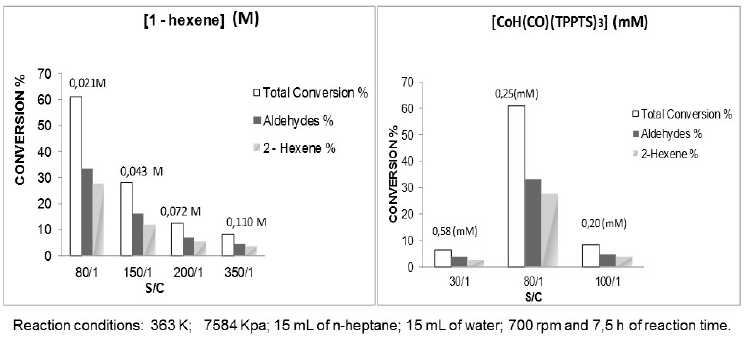
Fig. 7
Effect of 1-hexene concentration (a) and catalyst concentration (b) in the activity of the complex HCo(CO)(o-TPPTS)3.
In the case of the variation of the catalyst concentration Fig.7b, a maximum is observed at S/C ratio 80:1; when the concentration increase until 0.58 mM (S/C ratio 30/1), the catalytic precursor is to concentrated in the reaction medium and have difficulties for reached the active specie, which is confirmed by the increase of the induction time from 30 to 325 min (Table 2). In the case of 0.20 mM (S/C ratio 100/1), the catalytic active specie is too diluted, the total conversion decrease and the evolution of the reaction is to slow with an induction time of 175 min (Table 2).
Inductions time for the experiments of substrate - catalyst ratios.

Fig. 8 shows the conversion profile for 1-hexene and their products under the best operational conditions reaction. The l/b calculated ratio was 2.6, which is a very good value for lineal aldehyde. Finally, in order to confirm that the hydroformylation reaction proceeds in a homogeneous phase, the well-known mercury test [22,23] was carried out in several independent experiments and the results showed no interference at all with the evolution of the reaction and the hydroformylation carried on thought a molecular specie.
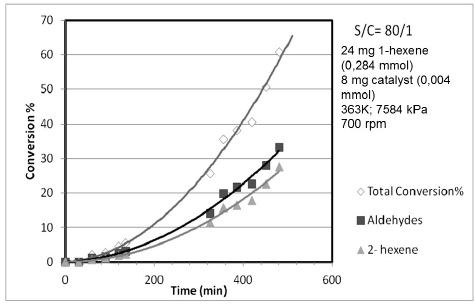
Fig. 8
Best condition for the catalytic activity of the complex HCo(CO)[P(o-C6H4SO3Na)]3.
Catalyst recycling
A very important aspect in biphasic catalysis is the capability of reusing the catalyst precursor for a long period of time without major changes in the nature, activity and selectivity [24]. Taking into account the above, a set of recycling experiments using the complex HCo(CO)[P(o-C6H4SO3Na)]3 during the hydroformylation of 1-hexene were performed. In this sense, four consecutive runs, maintaining the same aqueous phase during the whole experiment and changed the organic phase for a fresh phase which contains 1-hexene. The results obtained after this set of experiments can be seen in Figure 9.
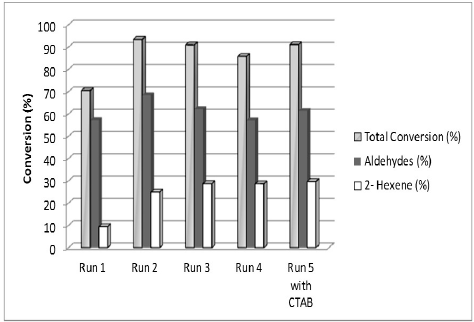
Fig. 9
Recycling of the catalyst precursor, HCo(CO)[P(o-C6H4SO3Na)]3 in the biphasic hydroformylation of 1- hexene.
As shown in Fig. 9, this complex efficiently catalyzed the hydroformylation of 1-hexene after four consecutive experiments (runs 2 - 5) without major indication of reducing the activity. Also in the run 5, a transfer agent such as cetyltrimethyl ammonium bromide CTAB, (5 mM) was added to the aqueous phase to improve the miscibility of catalyst precursor towards interface, the zone where the catalysis take place. Non-changes in the conversion, activity or selectivity of the system were observed. This experiment demonstrates the recycling capabilities of the catalytic precursor cobalt complex for the olefin hydroformylation reaction as well as the complex is close enough to the interface to perform effectively the catalysis.
Conclusion
The ligand tri(o-sulfonatophenyl)phosphine was synthesized as well as the cobalt complex HCo(CO)[P(o-C6H4SO3Na)]3 were prepared and fully characterized using different spectroscopy and mass spectrometry techniques. By proper tuning the condition for the sulfonation of the phosphine ligand, the sulfonate group was efficiently directed at ortho position. The great solubility in water of the complex allows use it for the first time as catalyst precursor in the biphasic hydroformylation reaction of 1-hexene under mild reaction conditions. During the 1-hexene hydroformylation, the increase of the syngas pressure improved the activity and selectivity to the aldehydes and the increment in temperature, increased the isomers production. The increment in the substrate concentration decrease the total conversion of the reaction and the increment in the catalyst concentration, reached a maximum at 0.25 mM at 363 K(90oC) and 7584 kPa (1100 psi) with more than 62% of olefin conversion and 66% selectivity towards aldehydes with l/b ratio =2.6. The hydride complex was recycled four successive times without lost in the activity, and maintaining the selectivity towards aldehydes around 60 %, which open the possibility for their use in industrial catalytic hydroformylation process.
Experimental section
General procedures
All manipulations were carried out under nitrogen atmosphere using standard Schlenck technique [25]. All the organic solvents were dried and purified by distillation and stored under inert atmosphere. The olefin and the respective saturated product were reagent grade (Aldrich Chemical). o-TPPTS was prepared according to modified reported procedures [21]. Water was distillated and saturated with nitrogen prior to use. Gases were purchased from Venezuela AGA-Gases. Infrared spectra were recorded in a Perkin Elmer Spectrum 1000 FTIR using samples as KBr disks. 1H and 31P{1H} NMR spectra were recorded on a Bruker 500 MHz spectrometer, using deuterated solvents. All chemical shifts are reported in parts per million (δ) relative to tetramethylsilane (1H) or 85% H3PO4 (31P). GC analyses were performed on a Hewlett Packard 5971 Pluss Series II chromatograph with a flame ionization detector in an ultra-1, DB-1 (10% dimethyl polixilosane) 25 m, 0.32 mm, 0.52 µm column to separate the products. Quantification was achieved by using n-decane as the internal standard and all peaks were identified by GC/MS on a Digital Technology 5890/5971 coupled system using a Quadrex PONA 10% dimethyl polyxilosane 25 m, 0,52 µm.
Synthesis and characterization of trisodium salt of 2,2´,2´´-phosphinetriylbenzene-sulfonic acid (o-TPPTS)
The TPPTS tri(o-sulfonatophenyl)phosphine was prepared according to the modified methods reported in the literature [20]. Over a period of 3h, 10.5 g sample of Triphenyl phosphine (TPP) was added slowly to 49 mL of 20 wt. % fuming sulfuric acid at 273K (0oC) and a further 16 mL of 20wt % is added with stirring. Efficient cooling is necessary to prevent local overheating. The reaction mixture was allowed to reach 278 - 283 K (5-10 oC) and the reaction continued for up to ca. 150 h to give complete sulfonation. The final reaction mixture was added in to the beaker containing 300 g of ice (using distillated and deoxygenated water). During the addition the internal temperature was kept 293 - 313 K (20-40oC). The mixture was stirred with a solution of 47.7 mL of triisooctylamine and 180 mL of toluene for 30 minutes and left to separate for additional 30 minutes. The aqueous phase was separated and discarded. The organic phase was treated twice with 5 wt. % aqueous sodium hydroxide. Firstly, base solution was added until the solution reaches a pH 5.5 and the aqueous solution was discarded. Secondly, the base solution was added to reaches a pH of 7.0 and the organic phase was discarded. The aqueous phase was rote-evaporated to dryness and subsequently washed with 500mL of a solution of acetone/methanol/water (10:5:1). (Yield 53 %)
1H NMR (500 MHz, D2O, δ ppm); 7.36 (1H; t); 7.44 (1H, t); 7.70 (1H, d); 7.77 (1H, d). 31P NMR (500 MHz, D2O, δ ppm); -5.76 (1P, s). 13C NMR (500 MHz, D2O, δ ppm);126,6 (1C, s); 129.7 (1C, s); 130,3 (1 C, d); 136.5 (2C, t); 143.1 (1 C, d). IR-FT (KBr; cm-1): υ (SO) 1208.93(st); 1149.46 (st); 1101.21(st); υ (OH) 3466.89 (st ). MS (MeOH, ESI): m/z: 522.84 [TPPTS - 2Na+]; 261,01 [PC18H14].
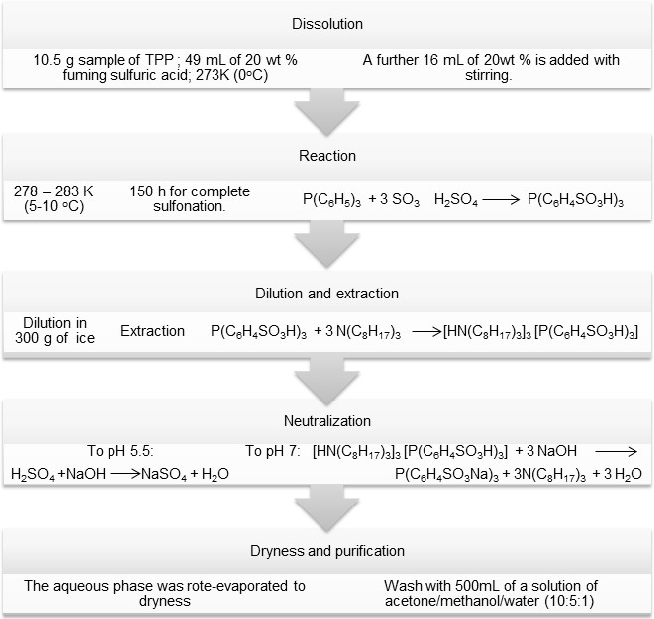
Scheme 2
Synthesis of trisodium salt of 2,2´,2´´-phosphinetriylbenzene-sulfonic acid (o-TPPTS).
Synthesis of HCo(CO)[P(o-C6H4SO3Na)]3
The hydridocarbonyltris[(o-sulfophenyl)phosphine)]rhodium(I) complex was synthesized according to the modified method reported by Hermann et al. [19] in which the purification of the complex using an ionic interchange resin was substituted by several cooled ethanol washed. In a 50 mL three neck round flask was introduced 500 mg (0.88 mmol) of TPPTS under three cycles of vacuum and nitrogen. Immediately 60 mg (0.25 mmol) of CoCl2.6H2O in 3 ml of distillated and de-oxygenated water were added to the phosphine. When the solution was completely homogeneous, the temperature was reduced to 273 K (0oC) using a bath ice and a solution of 16 mg of NaBH4 in 10 mL of de-oxygenated water was slowly added in drops approximately one-hour time. Simultaneously a current of carbon monoxide is passed through the solution during 6 h. After this time the ice bath and the current of CO are eliminated from the system. The obtained yellow solution was dried in the vacuum and washed three times with 10 mL portions of cooled ethanol. Finally, a dark green powder was obtained. (Yield: 0.37g; 85%)
1H NMR (500 MHz, D2O, δ ppm); 7.49 (36H; m); -12.466 (1H, q, J(P,H)42.6 Hz). 31P NMR (500 MHz, D2O, δ ppm); -5.76 (1P, s); 57.313 (3P, s); 34.19 (1P, s). 13C NMR (500 MHz, D2O, δ ppm); 126,5 (1C, s); 128.6 (1C, d); 129.64 (1C, d); 130.15(1C, d); 130.25 (1C, d); 134.1 (1C, d); 136.45(4C, q); 142.98 (1C,d); 143.60 (1C, d). IR-FT (KBr; cm-1): υ(CoH) 1955 (st); υ(CO) 1904.55 (sst); υ(SO) 1198(st); 1146 (st); 1098(st); υ (OH) 3451 (st ). MS (MeOH, ESI): m/z: [M - (o-TPPTS)] 1086,64; [TPPTS- 3Na+] 500.87.
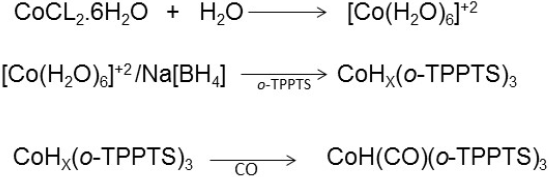
Scheme 3
Synthesis of HCo(CO)[P(o-C6H4SO3Na)]3.
Biphasic reaction
The catalytic runs were carried out under complete mixing which required high stirring speed of 750 rpm in our reactor. The experiments were conducted in a glass-lined stainless steel autoclave (Parr Instruments, 100 mL) which was coupled to a high pressure line and was provided with arrangements for sampling of liquid contents, automatic temperature and pressure control unit and variable stirrer speed. In a typical experiment, the catalyst precursor HCo(CO)[P(o-C6H4SO3Na)]3 (10 mg, 0.0056 mmol) in 15 mL of de-oxygenated water and 1-hexene (24 mg, 0.28 mmol) in 15 mL of heptane were introduced into the reactor. The solution was purged three times with CO/H2 (1:1, 500 kPa), then it was charged to the corresponding psi of syngas and heated to the adequate temperature (K). Samples of the reaction mixture were periodically extracted via a sampling valve, the total pressure was adjusted via a high pressure lung coupled to the reactor and the temperature keeps up constant by the temperature control unit. The samples were cooled, the phases separated and the organic phase analyzed by GC. The results of the catalytic reactions were analyzed by triplicated.
Recycling experiments:
The first run was made following the same method used for biphasic reactions. For the others: the aqueous phase from the last experiment was separate using a separation funnel and recycling with 24 mg (0.28mmol) in 15 mL of n-heptane. The solution was purged three times with CO/H2 (1:1, 5 atm), then it was charged to 7584 kPa (1100 psi) of syngas and heated to 363 K (90oC). Each experiment was conducted for 7.5h without collecting samples. At the end, the reactor was cooled, the phases separated and the organic phase analyzed by GC-FID.
Acknowledgements
We thank FONACIT-VENEZUELA for their financial support project F-97003766 and CDCH-UC project 94017. We are grateful to Universidad de Carabobo for permitting the publication of this work.
References
1.. 1. Kuntz E. G. CHEMTECH 1987, 17, 570.
2.. 2. Cornils B., Wiebus E., CHEMTECH, 1999, 33.
3.. 3. Cornils B., Kuntz E., Journal of Organometallic Chemistry. 1995, 502, 177.
4.. 4. Cornils B., Hermann W.A., Eckl R.W., Journal Molecular Catalysis 1997, 116, 27.
5.. 5. Cornils B., Hermann W.A., Aqueous Phase Organometallic Catalysis. Concept and Application, Wiley/VCH, Germany, 1998.
6.. 6. Van Leeuwen P.W.N.M., Claver C., in: James B.R., Van Leeuwen P.W.N.M. (Eds.), Rhodium Catalyzed Hydroformylation, Catalysis by Metal Complexes Series, vol. 22, Kluwer Academic Publishers, Dordrecht, 2000.
7.. 7. Cornils B., Herrmann W.A., in: Cornils B., Herrmann W.A. (Eds.), Applied Homogeneous Catalysis with Organometallic Compounds, 2nd ed., Wiley/VCH, Weinheim, 2002, 603-633 (Chapter.1.1.1).
8.. 8. Cornils B., Herrmann W.A. (Eds.), Aqueous Phase Organometallic Catalysis. Concepts and Applications, vol. 2, Wiley/VCH, Weinheim, 1998.
9.. 9. Joo F., Aqueous Organometallic Catalysis. Kluwer Academic Publishers, Dordrecht, 2001.
10.. 10. Desset S.L., Cole-Hamilton D.J., Foster D.F., Chemical Communications. 2007, 1933-1935.
11.. 11. Goedheijit M.S., Hanson B.E., Reek J.N.H., Kamer P.C.J., Van Leeuwen P.W.N.M. , Journal of American Chemical Society. 2000, 122, 1650-1657.
12.. 12. Monflier E., Tilloy S., Fremy G., Castanet Y., Mortreux A., Tetrahedron Lett. 1995, 36, 9481-9484.
13.. 13. Wiebus Ernst and Cornils B. in: Lindström Marcus U. Organic Reactions in Water. 2007, Blackwell Publishing; Chapter 12, 381-420.
14.. 14. Behr A. and Neubert P. Applied Homogeneous Catalysis. 2012, Wiley - VCH, Germany, 278-281.
15.. 15. Beller M.; Krauter J. G. E. Journal of Molecular Catalysis A: Chemical, 1999, 143, 31-39.
16.. 16. Parmar D. U. , Bajaj H. C.; Jasra R. V., Morosb B.M., Likholobov A. Journal of Molecular Catalysis A: Chemical. 2004, 211, 83-87.
17.. 17. Dabbawala A., Parmar D., Bajaj H. y Jasra R. Journal of Molecular Catalysis A: Chemical. 2008, 282, 99-106.
18.. 18. Baricelli P.J., Pacheco C., Vázquez A, Melean L. G., Modroño-Alonso M., Rosales M., González A. y Pacheco I. 2014 CIENCIA 22 (Número Especial), 104-111.
19.. 19. Herrmann W., Kellner J. and Riepl H. Journal of Organometallic Chemistry, 1990, 389 103-128.
20.. 20. (a) Bartik T.; Berit B.; Hanson B. E.; Glass T.; Bebout W. Inorganic Chemistry, 1992, 31, No. 12, 2667-2670. (b) Herrmann W. ; Kohlpaintner C. Inorganic Syntheses, 1998, 32, 8- 25.
21.. 21. Bartik T.; Bartik B.; Hanson B.; Whitmire K.; Guo I.; Inorganic Chemistry, 1993, 32, 5833-5837.
22.. 22. Anton D. R. and Crabtree R. Organometallics, 1983, 2, 855-859.
23.. 23. Lin Y. and Finke R. G. Inorganic Chemistry, 1994, 33, 4891-4910.
24.. 24. Cornils, B. and Wiebus, E., In: Cole-Hamilton, D.J. and Tooze, R.P. (Eds.), Catalyst Separation, Recovery and Recycle, Springer, London, 2006, 105.
25.. 25. Shriver D.F. The Manipulation of Air Sensitive Compounds, McGraw-Hill, USA, 1969.
Suplementary material:Author notes
*Corresponding author. Tel.: +58 412 3413286. E-mail address: [email protected]; [email protected]
